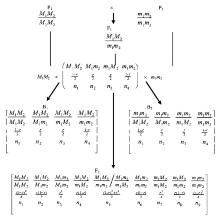
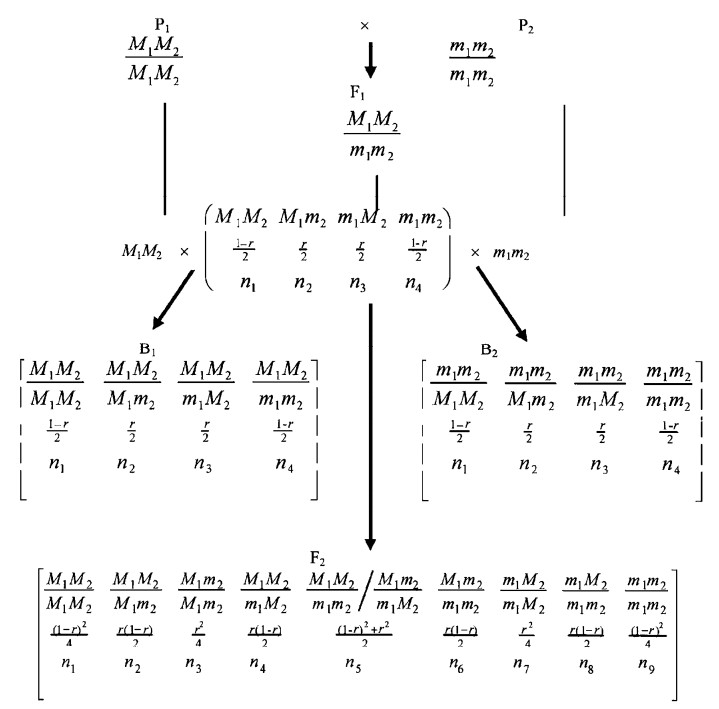
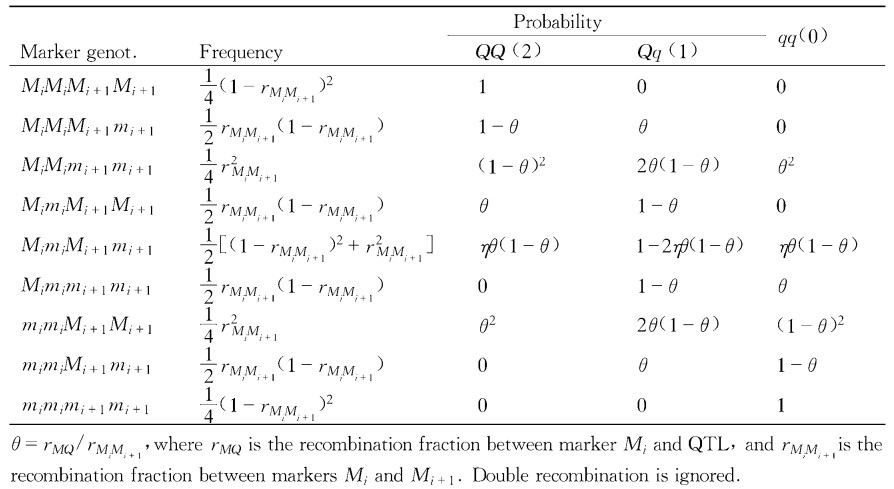
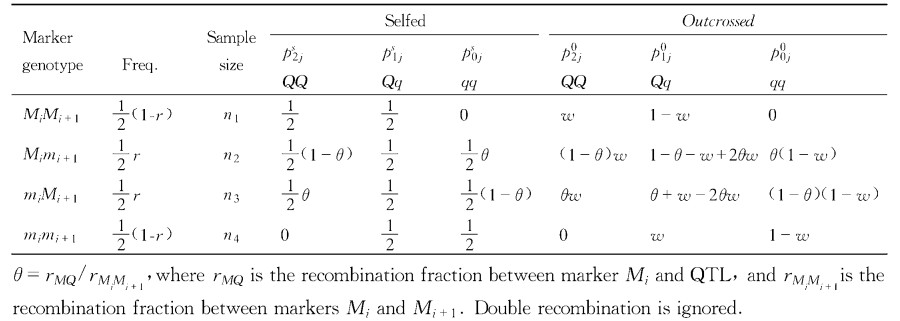
|
Benelli C , Bucci G . A genetic linkage map of Picea abies Karst, based on RAPD markers as a tool in population genetics Theor Appl Genet, 1994. 88, 283- 288.
|
|
Bierhorst DW . Morphology of vascular plants. New York: Macmillan. 1971.
|
|
Bradshaw HD Jr , Stettler RF . Molecular genetics of growth and development in Populus. Ⅳ. Mapping QTLs with large effects on growth, form and phenology in a forest tree Genetics, 1995. 139, 963- 973.
|
|
Broman KW 1997 Identifying quantitative trait loci in experimental crosvses. Ph. D thesis, University of California, Berkeley.
|
|
Castle WE . An improved method of estimating the number of genetic factors concerned in cases of blending in heritance Science, 1921. 54, 541- 553.
doi: 10.1126/science.54.1405.541
|
|
Churchill GA , Doerge RW . Empirical threshold values for quantitative trait mapping Genetics, 1994. 138, 963- 971.
|
|
Cockerham CC 1963 Estimates of genetic variances. In: Hanson WD; Robinson HF (eds) Statistical genetics and plant breeding. Proc Natl Acad Sci-NAS Coucil Publ 982. pp 53~94
|
|
Cockerham CC . Modifications in estimating the number of genes for a quantitative character Genetics, 1986. 114, 659- 664.
|
|
Comstock RE , Enfield FD . Gene number estimation when multiplicative genetic effects are assumed-growth in four beetles and mice Theor Appl Genet, 1981. 59, 373- 379.
doi: 10.1007/BF00276452
|
|
Dale GT 1994 Genetic mapping in quantitative trait analysis in the Pinus elliotti Enghelm.×Pinus carribaea More, hybrid and in Pinus radiata Don. Ph.D. thesis, University of Queensland, Brisbane, Australia
|
|
Feingold E , Brown PO , Siegmund D . Gaussian models for genetic linkage analysis using complete high-resolution maps of identity by descent Am J Hum Genet, 1993. 53, 234- 251.
|
|
Grattapaglia D; Wilcox P et al 1991 A RAPD map of loblolly pine in 60 days. Paper presented at the International Society for Plant Molecular Biology International Congress, Tucson, Arizona
|
|
Grattapaglia D , Bertolucci FLG , Sederoff RR . Genetic mapping of QTLs controlling vegetative propagation in in Eucalyptus grandis and E. urophylla using a pseudo-testcross mapping strategy and RAPD markers Genetics, 1995. 144, 1205- 1214.
|
|
Grattapaglia D , Bertolucci FLG , Penchel R , Sederoff RR . Genetic mapping of quantitative trait loci controlling growth and wood quality traits in Eucalyptus grandis using a maternal half-sib family and RAPD markers Genetics, 1996. 144, 1205- 1214.
|
|
Groover A , Devy M , Fiddler T , Lee J , Megraw T , Mitchell-Olds T , Sherman Vujcic C , Williams C , Neale D . Identification of quantitative trait loci influencing wood specific gravity in an outbred pedigree of loblolly pine Genetics, 1994. 138, 1293- 1300.
|
|
Haldane JBS . The combination of linkage values and the calculation of distance between the loci of linked factors J Genet, 1919. 8, 299- 309.
|
|
Hamrick JL , Godt MJW , Sherman-Broyles SL . Factors influencing levels of genetic diversity in woody plant species New Forests, 1992. 6, 95- 124.
doi: 10.1007/BF00120641
|
|
Huang QQ , Tomaru T , Wang LH , Ohba K . Genetic control of isozyme variation in Masson pine, Pinus massoniana Lamb Silvae Genet, 1994. 43, 285- 292.
|
|
Johannsen W . Elemente der exakten Erblichkeitslehre Custav Fischer, Jena, Germany, 1909.
|
|
Jansen RC . Interval mapping of multiple quantitative trait loci Genetics, 1993. 135, 205- 211.
|
|
Jansen RC . Controlling the type Ⅰ and type Ⅱ errors in mapping quantitative trait loci Genetics, 1994. 138, 871- 881.
|
|
Jansen RC , Stam P . High resolution of quantitative traits into multiple loci via interval mapping Genetics, 1994. 136, 1447- 1455.
|
|
Jiang C , Zeng Z-B . Multiple trait analysis of genetic mapping for quantitative trait loci Genetics, 1995. 140, 1111- 1127.
|
|
Jiang C , Zeng Z-B . Mapping quantitative trait loci with dominant and missing markers in various crosses from two inbred lines Genetica, 1997. 101, 47- 58.
doi: 10.1023/A:1018394410659
|
|
Jordan AP (1997) Fusiform rust disease resistance and genomic mapping in loblolly pine. Master thesis, North Carolina State University, Raleigh, NC
|
|
Kao C-H , Zeng Z-B . General formulas for obtaining the MLEs and the asymptotic variance-covariance matrix in mapping quantitative trait loci when using the EM algorithm Biometrics, 1997. 53, 653- 665.
doi: 10.2307/2533965
|
|
Kearsey MJ , Hyne V . QTL analysis: a simple marker-regression' approach Theor Appl Genet, 1994. 89, 698- 702.
doi: 10.1007/BF00223708
|
|
Knapp SJ , Bridges WC , Birkes D . Mapping quantitative trait loci using molecular marker linkage maps Theor Appl Genet, 1990. 79, 583- 592.
doi: 10.1007/BF00226869
|
|
Kosambi DD . The estimation of map values from recombination values Ann Eugen, 1944. 12, 172- 175.
|
|
Lande R . The minimum number of genes contributing to quantitative variation between and within populations Genetics, 1981. 99, 541- 553.
|
|
Lander ES , Botstein D . Mapping Mendelian factors underlying quantitative traits using RFLP linkage maps Genetics, 1989. 121, 185- 199.
|
|
Lynch M , WaLsh B . Genetics and analysis of quantitative traits Sinauer Associates, Sunderland, MA, 1998.
|
|
Martinez O , Curnow RN . Estimating the locations and the size of the effects of quantitative trait loci using flanking markers Theor Appl Genet, 1992. 85, 480- 488.
doi: 10.1007/BF00222330
|
|
Meng X-L , Rubin DB . Maximum likelihood estimation via the ECM algorithm: a general framework Biometrika, 1993. 80, 267- 278.
doi: 10.1093/biomet/80.2.267
|
|
McPeek MS , Spped TP . Modeling interferencer in genetic recombination Genetics, 1995. 139, 1031- 1044.
|
|
Millar CI . A steep cline in Pinus muricata Evolution, 1983. 37, 311- 319.
doi: 10.1111/j.1558-5646.1983.tb05541.x
|
|
Morton NE . LODs past and present Genetics, 1995. 140, 7- 12.
|
|
Muranty H . Power of tests for quantitative trait loci detection using full-sib families in different schemes Heredity, 1996. 76, 156- 165.
doi: 10.1038/hdy.1996.23
|
|
Nelson CD , Nance WL , Doudrick RL . A partial genetic linkage map of slash pine (Pinus elliottii Enghelm. var. elliottii) based on random amplified polymorphic DNAs Theor Appl Genet, 1993. 87, 145- 151.
doi: 10.1007/BF00223758
|
|
O'Malley DM, Whetten R 1997 Molecular markers and forest trees. In: Caetano- Anolles G, Gresshoff PM (eds) DNA markers: Protocols, applications and overviews. John Wiley & Sons, New York, pp. 237~257
|
|
Ott J . Analysis of human genetic linkage. John Hopkins Univ Press. 1991.
|
|
Plomion C , Bahrman N , DurelC-E , O'Malley DM . Genomic mapping in Pinus pinaster (maritime pine) using RAPD and protein markers Heredity, 1995. 74, 661- 668.
doi: 10.1038/hdy.1995.90
|
|
Rebai A , Goffinet B , Mangin B . Approximate thresholds of interval mapping tests for QTL detection Genetics, 1994. 138, 235- 240.
|
|
Rebai A , Goffinet B , Mangin B . Comparing power of different methods of QTL detection Biometrics, 1995. 51, 87- 99.
doi: 10.2307/2533317
|
|
Rogers DL 1997 Inheritance of allozymes from seed tissues of the hexaploid gymnosperm, Sequoiasempervirens (D. Don) Edl.(coast redwood). Heredity 78: 166~175
|
|
Sax K . The association of size differences with seed-coat pattern and pigmentation in Phaseolus vulgaris Genetics, 1923. 8, 552- 560.
|
|
Speed TJ , McPeek MS , Evans SN . Robustness of the non-interference model for ordering genetic models Proc Natl Acad Sci USA, 1992. 89, 3103- 3106.
doi: 10.1073/pnas.89.7.3103
|
|
Soller M , Brody T , Genizi A . On the power of experimental designs for the detection of linkage between marker loci and quantitative loci in crosses between inbred lines Theor Appl Genet, 1976. 47, 35- 39.
doi: 10.1007/BF00277402
|
|
Thoday JM . Location of polygenes Nature, 1961. 191, 368- 370.
doi: 10.1038/191368a0
|
|
Tulserium LK , Glaubitz JC , Kiss G , Carlson J . Single tree genetic linkage mapping using haploid DNA from megagame- tophytes BioTechnology, 1992. 10, 686- 690.
|
|
Weir BS . Genetic Data Analysis Sinauer Assoc., Sunderland, MA, 1996.
|
|
Weller JI . Maximum likelihood techniques for the mapping and analysis of quantitative trait loci with the aid of genetic markers Biometrics, 1987. 42, 627- 640.
|
|
Wheeler NC , Guries RP . Population structure, genetic diversity, and morphological variation in Pinus contorta Dougl Can J For Res, 1982. 12, 595- 606.
doi: 10.1139/x82-091
|
|
Wilcox PL , Amerson HV , Kuhlman EG , Liu B-H , O'Malley DM , Sederoff RR . Detection of a major gene for resistance to fusiform rust disease in loblolly pine by genomic mapping Proc Natl Acad Sci, 1996. 93, 3859- 3864.
doi: 10.1073/pnas.93.9.3859
|
|
Wright AJ , Mowers RP . Multiple regression for molecular-marker, quantitative trait data from large F2 populations Theor Appl Genet, 1994. 89, 305- 312.
|
|
Wu RL . Quantitative-genetic dissection of complex traits in a QTL-mapping pedigree Theor Appl Genet, 1996. 93, 447- 457.
doi: 10.1007/BF00223189
|
|
Wu RL . Genetic mapping of QTLs affecting tree growth and architecture in Populus: implications for ideotype breeding Theor Appl Genet, 1998a. 96, 447- 457.
doi: 10.1007/s001220050761
|
|
Wu RL . Mapping quantitative trait loci by genotyping haploid tissues. Genetics, in press. 1998b.
|
|
Wu RL . Molecular evidence for epigenetic inheritance. Dev Genets in press. 1998c.
|
|
Wu WR , Li WM . A new approach for mapping quantitative trait loci using complete genetic marker linkage maps Theor Appl Genet, 1994. 89, 535- 539.
doi: 10.1007/BF00222444
|
|
Xu S , Atchley WR . A random model approach to interval mapping of quantitative trait loci Genetics, 1995. 141, 1189- 1197.
|
|
Yan T 1997 Construction of Genetic Linkage maps in Populus and Pinus massoniana. Ph. D. thesis, Nanjing Forestry University, Nanjing, China
|
|
Yazdani R , Yeh FC , Rimsha J . Genomic mapping of Pinus sylvestris (L ) using random amplified polymorphic DNA markers. Forest Genet, 1995. 2, 109- 116.
|
|
Zeng Z-B . Correcting the bias of Wright' s estimates of the number of genes affecting a quantitative character-A further improved method Genetics, 1992. 131, 987- 1001.
|
|
Zeng Z-B . Theoretical basis of precision mapping of quantitative trait loci Proc Natl Acad Sci USA, 1993. 90, 10972- 10976.
doi: 10.1073/pnas.90.23.10972
|
|
Zeng Z-B . Precision mapping of quantitative trait loci Genetics, 1994. 136, 1457- 1568.
|
|
Zhao H , McPeek MS , Speed TP . Statistical analysis of chromatid interference Genetics, 1995a. 139, 1057- 1065.
|
|
Zhao H , Speed TP , McPeek MS . Statistical analysis of crossover interference using the chi-square model Genetics, 1995b.139, 1045- 1056.
|