
Scientia Silvae Sinicae ›› 2021, Vol. 57 ›› Issue (9): 110-120.doi: 10.11707/j.1001-7488.20210911
Previous Articles Next Articles
Xingzhou Chen,Guoying Zhou,Xinggang Chen,Lingyu Jiang,Anhua Bao,Jun Liu*
Received:2020-09-01
Online:2021-09-25
Published:2021-11-29
Contact:
Jun Liu
CLC Number:
Xingzhou Chen,Guoying Zhou,Xinggang Chen,Lingyu Jiang,Anhua Bao,Jun Liu. Screening of Effectors of Colletotrichum fructicola in Camellia oleifera[J]. Scientia Silvae Sinicae, 2021, 57(9): 110-120.
Table 1
9 candidate effectors gene-specific primer sequences designed for qRT-PCR"
| 基因ID GenBank ID | 扩增长度Amplification length/bp | 正向引物Forward primer | 反向引物Reverse primer |
| CGGC5_3865 | 193 | TTCTCCCTCCAGACCATCG | TGCAGACGTCCTGCTTCAA |
| CGGC5_5363 | 185 | TGCCCGTGGATACTGAACAA | ATCCTTTGGCACTTCCTGCT |
| CGGC5_7791 | 96 | CAAGGGCAACGGTCTCTTCT | CTGGGATCGTTGGCGTTTTC |
| CGGC5_398 | 114 | CTCTACCCTCCTCACCGTCT | GAGTTGTTGCAGTGCTTCAG |
| CGGC5_10060 | 122 | GCCGCCTCTTCCAATGCT | AAGTCCCCGTGTTGACTTC |
| CGGC5_7871 | 65 | CACCTACTGCGACCCCTACTC | CCTTCTCAACACAGGCTCTGG |
| CGGC5_462 | 409 | GCCTTTTTTGCCCTTTTGCTG | CAGGCTGTTCTTGTTGTCGGC |
| CGGC5_15187 | 156 | TTTGACTGCAAGTGCAAGGACGAT | ACCACAGCAGGTCTTGAACGGGAA |
| CGGC5_7737 | 64 | TCCGACTGCCACGACAACTA | TGCATCCAGAACGCTGAAGA |
Table 2
4 candidate effector gene-primer sequences designed for full-length cloning"
| 基因ID(编号) GenBank ID(number) | 扩增长度Amplification length/bp | 正向引物Forward primer | 反向引物Reverse primer |
| CGGC5_3865(CfEP1) | 348 | ATGAAGTTCTCCCTCCAGACCAT | CTAGGCGCCAGGGGTGC |
| CGGC5_7871(CfEP2) | 282 | ATGAAGGCTTTCACCGTCC | TTGGTGCACTTGCCATCC |
| CGGC5_15187(CfEP3) | 243 | ATGCAGATTCAGAACGTTGTCC | TTATCGGCACAAGTCGAAGC |
| CGGC5_7737(CfEP4) | 207 | ATGCTCTTCTCCAAGACCATCC | TTAGCAGTCGTCGCACATGA |
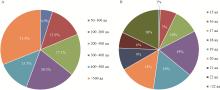
Fig.1
Characteristic analysis of classic secreted proteins A: Amino acid length analysis; B: Signal peptide length analysis."
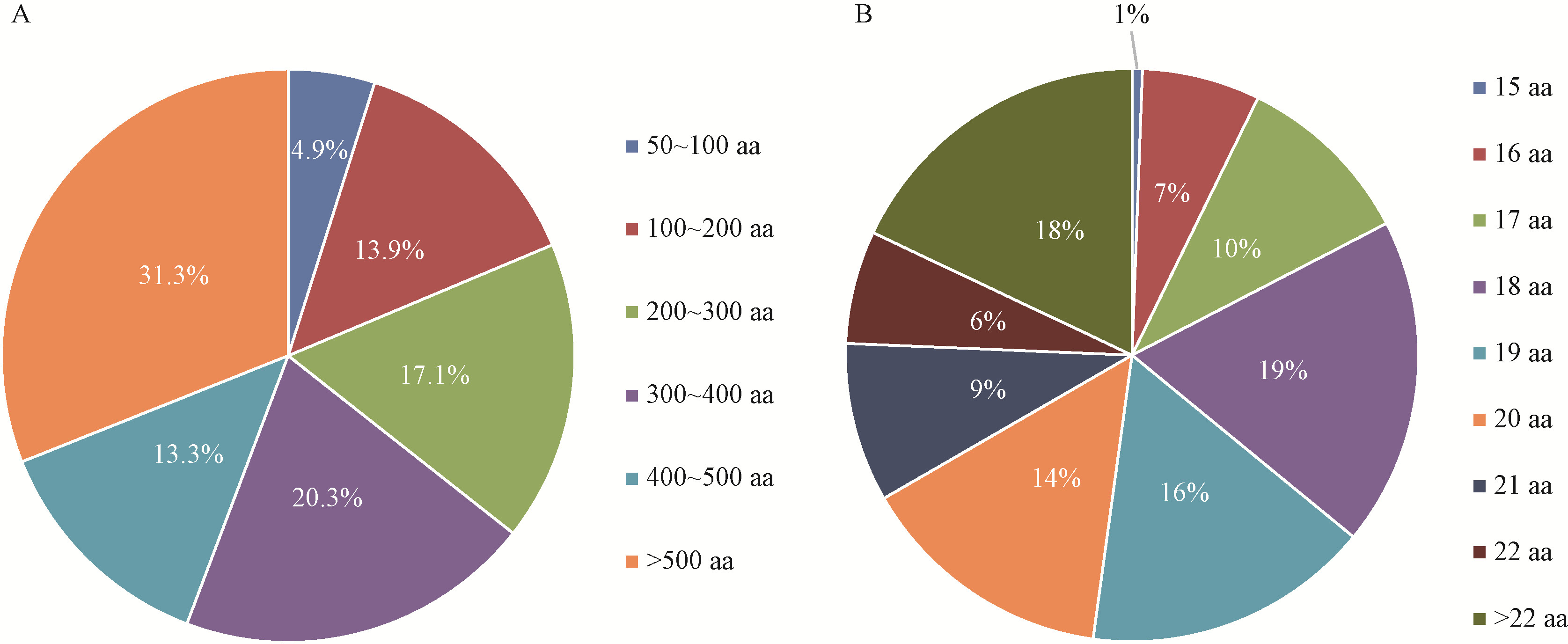
Fig.2
The ratio of amino acids in C. fructicola classic secreted proteins"
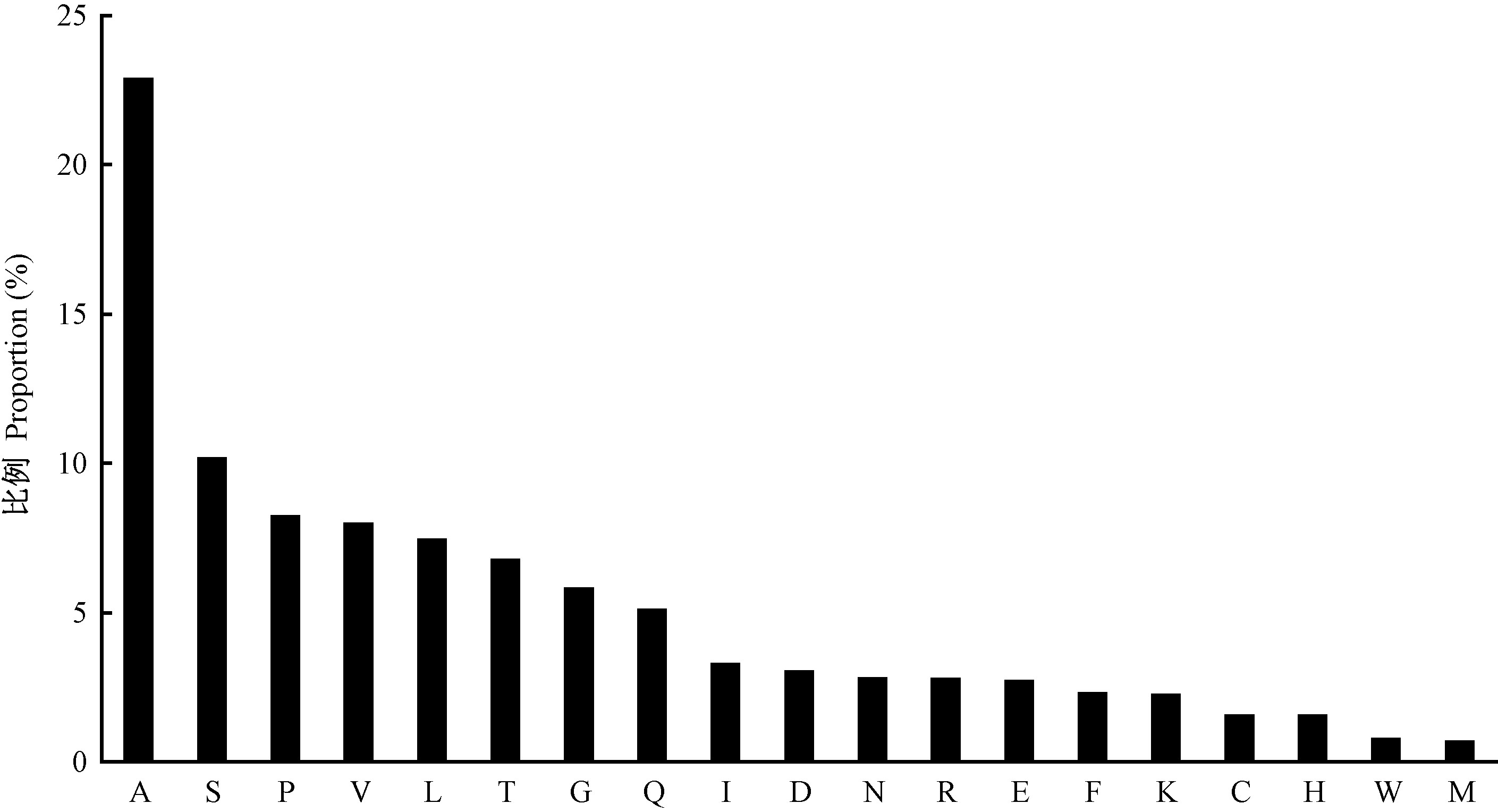

Fig.3
GO classifications of the classic secreted proteins inC. fructicola A: Molecular function; B: Biological process; C: Cellular component."

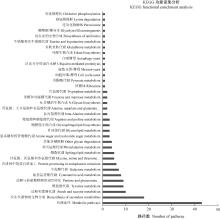
Fig.4
KEGG classifications of the classically secreted proteins in C. fructicola"
Table 3
31 Pathogenic related gene information"
| 基因ID GenBank ID | 基因名称Gene name | 突变类型Mutant phenotype | E-value | 分数Score | 功能Function |
| CGGC5_14282 | SLP 1 | 效应物Effector | 1.17E-57 | 175 | 效应蛋白Effector protein |
| CGGC5_14341 | SPM1 | 毒力降低Reduced virulence | 4.52E-64 | 210 | 蛋白酶活性Protease activity |
| CGGC5_14887 | bcpme1 | 毒力降低Reduced virulence | 1.83E-74 | 240 | 果胶甲酯酶Pectin methylesterase |
| CGGC5_15366 | SPM1 | 毒力降低Reduced virulence | 2.41E-69 | 225 | 蛋白酶活性Protease activity |
| CGGC5_1639 | Ss-ggt1 | 毒力降低Reduced virulence | 1.94E-116 | 352 | γ-谷氨酰转肽酶Gamma-glutamyl transpeptidase |
| CGGC5_1824 | PELA | 毒力降低Reduced virulence | 2.56E-62 | 196 | 果胶裂解酶Pectate lyase |
| CGGC5_198 | MGG_10510 | 毒力降低Reduced virulence | 4.64E-116 | 335 | 假定蛋白Hypothetical protein |
| CGGC5_243 | GAS1 | 毒力降低Reduced virulence | 6.19E-110 | 314 | 渗透Appressorial penetration |
| CGGC5_2526 | SPM1 | 毒力降低Reduced virulence | 3.94E-60 | 202 | 蛋白酶活性Protease activity |
| CGGC5_3265 | pnl1 | 毒力降低Reduced virulence | 5.10E-127 | 367 | 果胶裂解酶Pectin lyase |
| CGGC5_3276 | CTB5 | 毒力降低Reduced virulence | 1.58E-43 | 157 | 氧化还原酶Oxidoreductase |
| CGGC5_3706 | CPA1 | 毒力降低Reduced virulence | 3.01E-70 | 211 | 亲环蛋白Cyclophilin |
| CGGC5_6227 | CMLE | 丧失致病性Loss of pathogenicity | 1.86E-62 | 201 | 羟基顺式、顺式粘康酸环化酶Carboxy-cis, cis-muconate cyclase |
| CGGC5_6331 | Gas1 | 丧失致病性Loss of pathogenicity | 0 | 740 | 内置网葡萄糖苷酶Endoplasmic reticulum glucosidase |
| CGGC5_6379 | IPP1 | 致死Lethal | 4.33E-71 | 224 | 无机焦磷酸酶Inorganic pyrophosphatase |
| CGGC5_669 | PELA | 毒力降低Reduced virulence | 3.63E-63 | 196 | 果胶裂解酶Pectate lyase |
| CGGC5_7177 | CTB5 | 毒力降低Reduced virulence | 7.97E-26 | 107 | 氧化还原酶Oxidoreductase |
| CGGC5_7352 | CTB5 | 毒力降低Reduced virulence | 4.86E-48 | 169 | 氧化还原酶Oxidoreductase |
| CGGC5_8160 | endo-1, 4-beta-xylanase | 毒力降低Reduced virulence | 0 | 527 | 内切-β-1,4-木聚糖酶Endo-β-1, 4-xylanase |
| CGGC5_8220 | pnl1 | 毒力降低Reduced virulence | 1.13E-122 | 356 | 果胶裂解酶Pectin lyase |
| CGGC5_9008 | lac2 | 毒力降低Reduced virulence | 2.23E-88 | 283 | 漆酶基因Laccase gene |
| CGGC5_9093 | XYN11A | 毒力降低Reduced virulence | 6.19E-39 | 132 | 内切-β-1,4-木聚糖酶Endo-β-1, 4-xylanase |
| CGGC5_9386 | CTB5 | 毒力降低Reduced virulence | 1.03E-26 | 110 | 氧化还原酶Oxidoreductase |
| CGGC5_9512 | SPM1 | 毒力降低Reduced virulence | 1.89E-54 | 186 | 蛋白酶活性Protease activity |
| CGGC5_11305 | MGG_04128 | 毒力降低Reduced virulence | 0 | 813 | 假定蛋白Hypothetical protein |
| CGGC5_11629 | PELA | 毒力降低Reduced virulence | 8.23E-71 | 215 | 果胶裂解酶Pectate lyase |
| CGGC5_13316 | FET3-1 | 毒力降低Reduced virulence | 2.10E-149 | 441 | 功能性铁氧化酶Functional ferroxidase |
| CGGC5_13439 | PELD | 毒力降低Reduced virulence | 3.23E-92 | 268 | 果胶裂解酶Pectate lyase |
| CGGC5_13646 | CYB2 | 毒力降低Reduced virulence | 4.52E-51 | 177 | 乳酸脱氢酶Lactate dehydrogenase |
| CGGC5_13947 | Avenacinase gene | 丧失致病性Loss of pathogenicity | 0 | 840 | 阿唯那霉素酶Avenacinase |
| CGGC5_14187 | Avenacinase gene | 丧失致病性Loss of pathogenicity | 6.17E-45 | 156 | 阿唯那霉素酶Avenacinase |
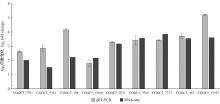
Fig.5
Quantitative analysis of nine candidate effectors from C. fructicola transcriptome Comparison of RNA-Seq and qRT-PCR validation results. Error bars represent the standard error of the mean."


Fig.6
Functional verification of 4 candidate effectors in C. fructicola"

| 何燕华. 2014. 稻瘟菌新型效应蛋白的筛选及功能初步分析. 福州: 福建师范大学硕士学位论文. | |
| He Y H. 2014. Identification and preliminarily functional analysis of new effector proteins from Magnaporthe oryzae. Fuzhou: MS thesis of Fujian Normal University. [in Chinese] | |
| 李河. 2018. 油茶炭疽病菌群体遗传及MAPK基因CfPMK1功能研究. 长沙: 中南林业科技大学博士学位论文. | |
| Li H. 2018. Population genetic analyses of the fungal pathogen Colletotrichum on tea-oil trees in China and characterization of a MAPK gene CfPMK1 in the pathogen. Changsha: PhD thesis of Central South University of Forestry and Technology. [in Chinese] | |
| 李司政, 李河. 果生刺盘孢CfHAC1调控应答二硫苏糖醇胁迫的转录组分析. 菌物学报, 2020, 39 (10): 1886- 1896. | |
| Li S Z , Li H . Genome-wide transcriptome analysis of Colletotrichum fructicola CfHAC1 regulation of the response to dithiothreitol stress. Mycosystema, 2020, 39 (10): 1886- 1896. | |
|
喻锦秀, 聂云安, 周刚, 等. 湖南省油茶主要病害发生规律研究. 湖南林业科技, 2014, 41 (1): 94- 97.
doi: 10.3969/j.issn.1003-5710.2014.01.018 |
|
|
Yu J X , Nie Y A , Zhou G , et al. Occurrence regularity of main disease of Camellia oleifera in Hunan. Hunan Forestry Science & Technology, 2014, 41 (1): 94- 97.
doi: 10.3969/j.issn.1003-5710.2014.01.018 |
|
| 张丽勍, 段可, 邹小花, 等. 2016. 胶孢炭疽菌侵染草莓的转录组学研究. 中国植物病理学会2016年学术年会论文集. | |
| Zhang L Q, Duan K, Zou X H, et al. 2016. Transcriptomics study of Colletotrichum gloeosporioides infecting strawberry. Proceedings of the 2016 Annual Conference of the Chinese Society of Plant Pathology. [in Chinese] | |
|
Castrense S , Liugi M P , Piero F , et al. BUSCA: an integrative web server to predict subcellular localization of proteins. Nucleic Acids Research, 2018, 46 (W1): W459- W466.
doi: 10.1093/nar/gky320 |
|
|
Dangl J L , Horvat D M , Staskawicz B J , et al. Pivoting the plant immune system from dissection to deployment. Science, 2013, 341 (6147): 746- 751.
doi: 10.1126/science.1236011 |
|
|
Djamei A , Schipper K , Rabe F , et al. Metabolic priming by a secreted fungal effector. Nature, 2011, 478 (7369): 395- 398.
doi: 10.1038/nature10454 |
|
|
Emanuelsson O , Brunak S , Heijne G , et al. Locating proteins in the cell using TargetP, SignalP and related tools. Nature Protocols, 2007, 2 (4): 953- 971.
doi: 10.1038/nprot.2007.131 |
|
|
Emanuelsson O , Nielsen H , Brunak S , et al. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. Journal of Molecular Biology, 2000, 300 (4): 1005- 1016.
doi: 10.1006/jmbi.2000.3903 |
|
|
Halaouli S , Asther M , Sigoillot J C , et al. Fungal tyrosinases: new prospects in molecular characteristics, bioengineering and biotechnological applications. Journal of Applied Microbiology, 2006, 100 (2): 219- 232.
doi: 10.1111/j.1365-2672.2006.02866.x |
|
|
Jana S , Peter N D , Donald M G , et al. Improved prediction of fungal effector proteins from secretomes with Effector P2.0. Molecular Plant Pathology, 2018, 19 (9): 2094- 2110.
doi: 10.1111/mpp.12682 |
|
|
Jannick D B , Nielsen H , von Heijne G , et al. Improved prediction of signal peptides: signal P3.0. Journal of Molecular Biology, 2004, 340 (4): 783- 795.
doi: 10.1016/j.jmb.2004.05.028 |
|
| Jin Q , Dong H , Peng Y , et al. Application of cDNA array for studying the gene expression profile of mature appressoria of Magnaporthe grisea. Journal of Zhejiang University Science B: Biomedicine & Biotechnology, 2007, 8 (2): 88- 97. | |
|
Jones D A , Bertazzoni S , Turo C J , et al. Bioinformatic prediction of plant-pathogenicity effector proteins of fungi. Current Opinion in Microbiology, 2018, 46, 43- 49.
doi: 10.1016/j.mib.2018.01.017 |
|
|
Jones J D G , Dangl J L . The plant immune system. Nature, 2006, 444 (7117): 323- 329.
doi: 10.1038/nature05286 |
|
|
Jonge R D , Esse H P V , Kombrink A , et al. Conserved fungal LysM effector Ecp6 prevents chitin-triggered immunity in plants. Science, 2010, 329 (5994): 953- 955.
doi: 10.1126/science.1190859 |
|
|
Juncker A S , Willenbrock H , Von Heijne G , et al. Prediction of lipoprotein signal peptides in gram-negative bacteria. Protein Science, 2003, 12 (8): 1652- 1662.
doi: 10.1110/ps.0303703 |
|
| Kanehisa M , Sato Y . KEGG mapper for inferring cellular functions from protein sequences. Protein Science, 2019, 29 (1): 28- 35. | |
|
Ke X , Yin Z , Song N , et al. Transcriptome profiling to identify genes involved in pathogenicity of Valsa mali on apple tree. Fungal Genetics and Biology, 2014, 68, 31- 38.
doi: 10.1016/j.fgb.2014.04.004 |
|
|
Kushalappa A C , Yogendra K N , Karre S . Plant innate immune response: qualitative and quantitative resistance. Critical Reviews in Plant Sciences, 2016, 35 (1): 38- 55.
doi: 10.1080/07352689.2016.1148980 |
|
|
Liang X F , Shang S P , Dong Q Y , et al. Transcriptomic analysis reveals candidate genes regulating development and host interactions of Colletotrichum fructicola. BMC Genomics, 2018, 19 (1): 557- 578.
doi: 10.1186/s12864-018-4934-0 |
|
| Martin U , Alayne C , James S , et al. PHI-base: the pathogen-host interactions database. Nucleic Acids Research, 2019, 48 (D1): D613- D620. | |
|
Peng T , Orsborn K I , Orbach M J , et al. Proline-rich vaccine candidate antigen of Coccidioides immitis: conservation among isolates and differential expression with spherule maturation. Journal of Infectious Diseases, 1999, 179 (2): 518- 521.
doi: 10.1086/314604 |
|
|
Ramachandran S R , Yin C , Kud J , et al. Effectors from wheat rust fungi suppress multiple plant defense responses. Phytopathology, 2017, 107 (1): 75- 83.
doi: 10.1094/PHYTO-02-16-0083-R |
|
|
Shang S P , Wang B , Zhang S , et al. A novel effector CfEC92 of Colletotrichum fructicola contributes to glomerella leaf spot virulence by suppressing plant defences at the early infection phase. Molecular Plant Pathology, 2020, 21 (7): 936- 950.
doi: 10.1111/mpp.12940 |
|
|
Sperschneider J , Dodds P N , Gardiner D M , et al. Advances and challenges in computational prediction of effectors from plant pathogenic fungi. PLoS Pathogens, 2015, 11 (5): e1004806.
doi: 10.1371/journal.ppat.1004806 |
|
|
Yan W , Wang Y . Trick or treat: microbial pathogens evolved apoplastic effectors modulating plant susceptibility to infection. Molecular Plant-Microbe Interactions, 2018, 31 (1): 6- 12.
doi: 10.1094/MPMI-07-17-0177-FI |
| [1] | Zijing Zhou,Fuhua Fan,Xianwen Shang,Huijuan Qin,Conghui Wang,Guijie Ding,Jianhui Tan. Effects of Exogenous IAA on Stem Secondary Growth of Pinus massoniana Seedlings [J]. Scientia Silvae Sinicae, 2021, 57(9): 42-51. |
| [2] | Peng Xin, Wang Hantang, Guo Chunhui, Yang Zhende, Zhou Jing, Wang Xue, Ding Zhirou. EST-SSR Development and Cryptic Species Identification of the Invasive Gall-Causing Pest Leptocybe invasa (Hymenopetra: Eulophidae) [J]. Scientia Silvae Sinicae, 2021, 57(9): 140-151. |
| [3] | Xiya Li,Shengpei Zhang,He Li. Function of Vacuolar Protein Sorting CfVps26 in Colletotrichum fructicola on Camellia oleifera [J]. Scientia Silvae Sinicae, 2021, 57(8): 94-101. |
| [4] | Jinfeng Cai,Xiaoming Yang,Wanwen Yu,Guibin Wang,Fuliang Cao. Development of SSR Molecular Markers Based on Transcriptome Sequencing of Melia azedarach [J]. Scientia Silvae Sinicae, 2021, 57(6): 85-92. |
| [5] | Yalan Gao,Yuanhao He,He Li. Biological Function bZIP-Type Transcription Factor CfAp1 in Colletotrichum fructicola [J]. Scientia Silvae Sinicae, 2020, 56(9): 30-39. |
| [6] | Peihuang Zhu,Yu Chen,Lingzhi Zhu,Rong Li,Kongshu Ji. Codon Usage Bias and Its Influencing Factors in Pinus massoniana Transcriptome [J]. Scientia Silvae Sinicae, 2020, 56(4): 74-81. |
| [7] | Xiaoyu Lu,Zhu Chen,Fei Tang,Songling Fu,Jie Ren. Combined Transcriptomic and Metabolomic Analysis Reveals Mechanism of Anthocyanin Changes in Red Maple(Acer rubrum) Leaves [J]. Scientia Silvae Sinicae, 2020, 56(1): 38-53. |
| [8] | Zhao Qingquan, Chi Yujie, Zhang Jian, Feng Lianrong. Transcriptome Construction and Related Gene Expression Analysis of Lenzites gibbosa in Woody Environment [J]. Scientia Silvae Sinicae, 2019, 55(8): 95-105. |
| [9] | Han Xiaohong, Lu Ciding, Hua Yin, Lin Haoyu, Shi Yufei, Wu Songqing, Zhang Feiping, Liang Guanghong. Phylogenetic Analysis of Transcriptome and Three Detoxification Enzyme Families Related Genes in Anoplophora chinensis (Coleoptera: Cerambycidae) [J]. Scientia Silvae Sinicae, 2019, 55(5): 104-113. |
| [10] | Zhang Enliang, Ma Lingling, Yang Rutong, Li Linfang, Wang Qing, Li Ya, Wang Peng. Transcriptome Profiling of IBA-Induced Adventitious Root Formation in Softwood Cuttings of Catalpa bungei ‘Yu-1’ [J]. Scientia Silvae Sinicae, 2018, 54(5): 48-61. |
| [11] | Mao Weibing, Chen Faju, Wang Changlan, Liang Hongwei. Transcriptome Sequencing and Analysis of Male Sterile Flower Buds in Catalpa bungei [J]. Scientia Silvae Sinicae, 2017, 53(6): 141-150. |
| [12] | Shi Xiaodong, Zhu Xuehui, Sheng Yuzhen, Zhuang Guoqing, Chen Fang. Development of SSR Markers Based on Transcriptome Sequence of Phoebe zhennan [J]. Scientia Silvae Sinicae, 2016, 52(11): 71-78. |
| [13] | Li He, Li Yang, Xu Jianping, Zhou Guoying. Population Genetic Structure of Colletotrichum fructicola from Oil-Tea and Other Host Plants in Hainan province [J]. Scientia Silvae Sinicae, 2016, 52(10): 80-88. |
| [14] | Zhang Zhen, Zhang Hanguo, Mo Chi, Zhang Lei. Transcriptome Sequencing Analysis and Development of EST-SSR Markers for Pinus koraiensis [J]. Scientia Silvae Sinicae, 2015, 51(8): 114-120. |
| [15] | Yin Liwei, Chi Yujie. Cloning and Bioinformatics Analysis of MnP 1 cDNA Gene from Hericium erinaceum [J]. Scientia Silvae Sinicae, 2015, 51(5): 68-77. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||