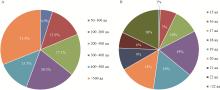
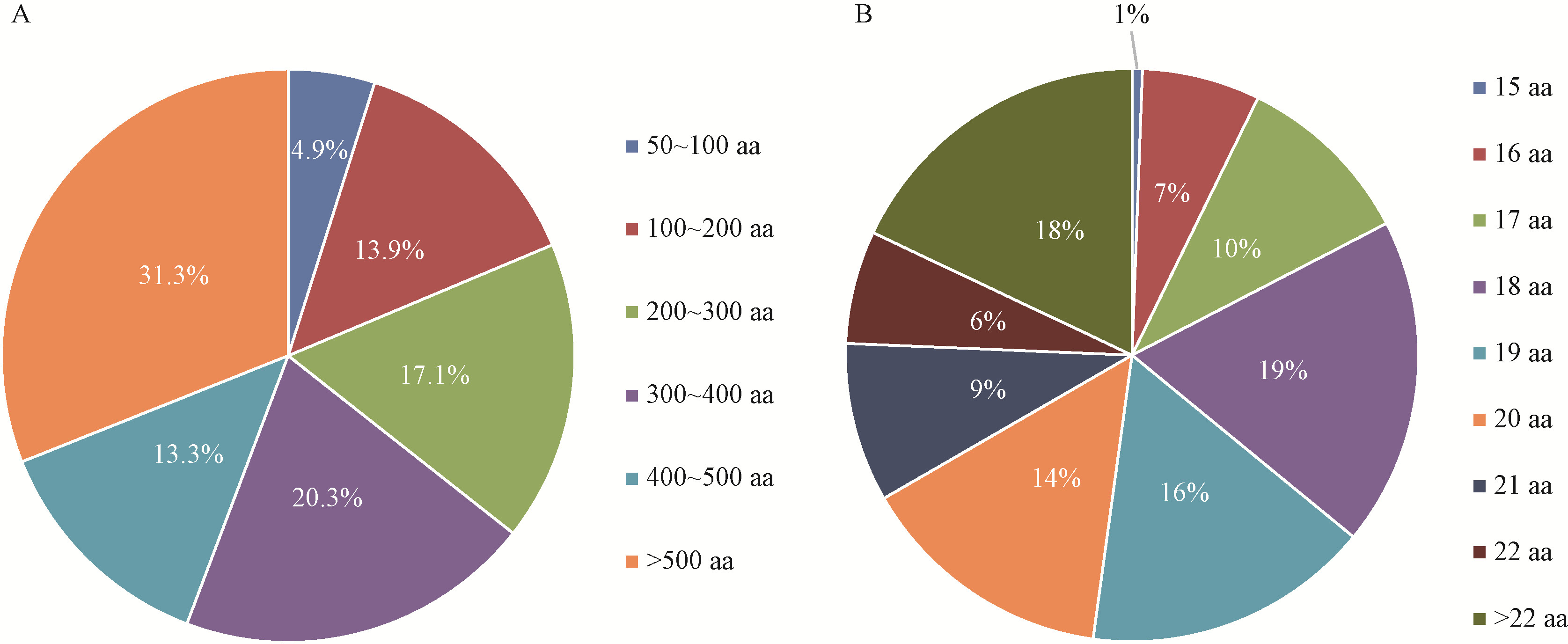
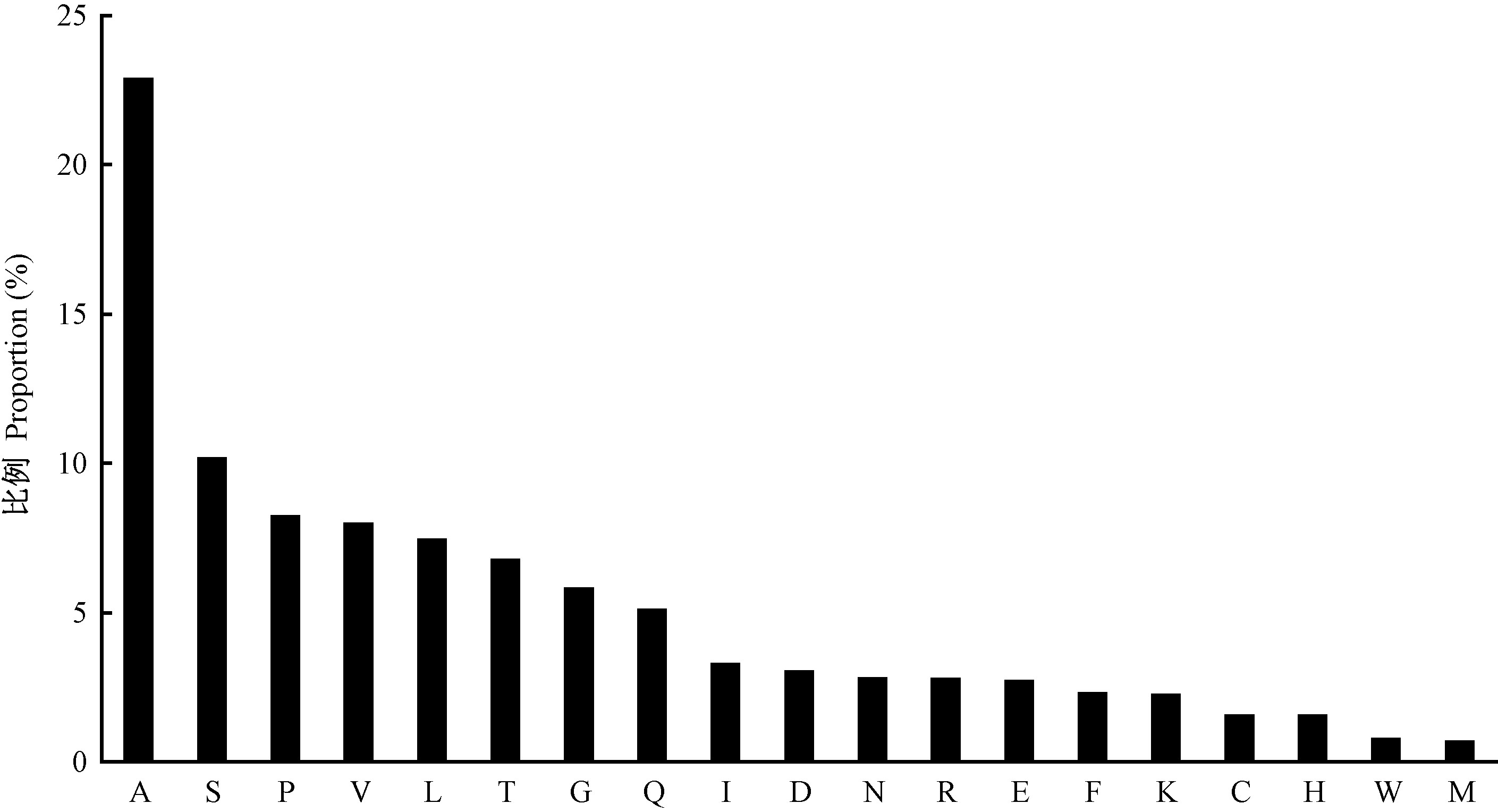
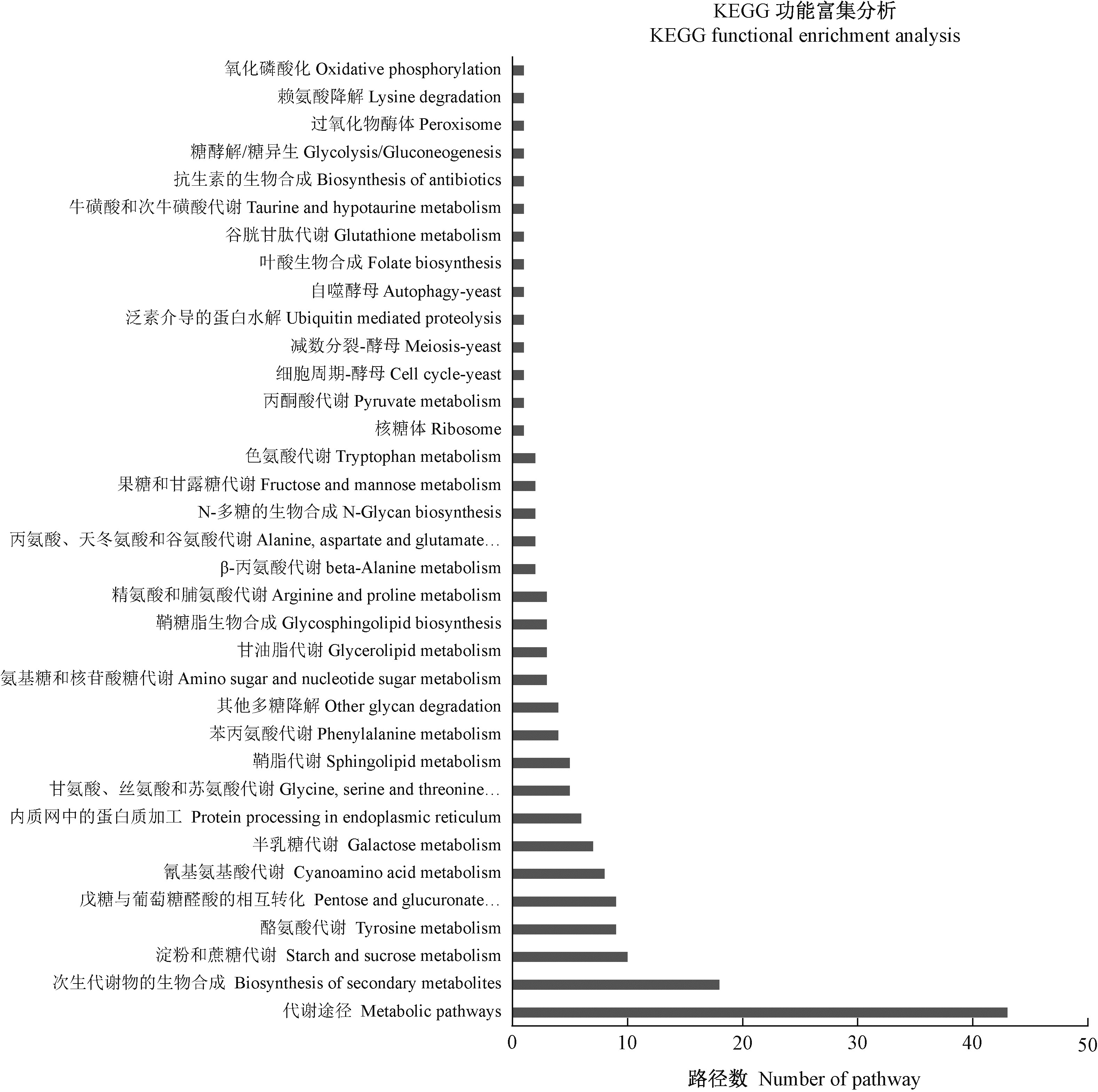
|
何燕华. 2014. 稻瘟菌新型效应蛋白的筛选及功能初步分析. 福州: 福建师范大学硕士学位论文.
|
|
He Y H. 2014. Identification and preliminarily functional analysis of new effector proteins from Magnaporthe oryzae. Fuzhou: MS thesis of Fujian Normal University. [in Chinese]
|
|
李河. 2018. 油茶炭疽病菌群体遗传及MAPK基因CfPMK1功能研究. 长沙: 中南林业科技大学博士学位论文.
|
|
Li H. 2018. Population genetic analyses of the fungal pathogen Colletotrichum on tea-oil trees in China and characterization of a MAPK gene CfPMK1 in the pathogen. Changsha: PhD thesis of Central South University of Forestry and Technology. [in Chinese]
|
|
李司政, 李河. 果生刺盘孢CfHAC1调控应答二硫苏糖醇胁迫的转录组分析. 菌物学报, 2020, 39 (10): 1886- 1896.
|
|
Li S Z , Li H . Genome-wide transcriptome analysis of Colletotrichum fructicola CfHAC1 regulation of the response to dithiothreitol stress. Mycosystema, 2020, 39 (10): 1886- 1896.
|
|
喻锦秀, 聂云安, 周刚, 等. 湖南省油茶主要病害发生规律研究. 湖南林业科技, 2014, 41 (1): 94- 97.
doi: 10.3969/j.issn.1003-5710.2014.01.018
|
|
Yu J X , Nie Y A , Zhou G , et al. Occurrence regularity of main disease of Camellia oleifera in Hunan. Hunan Forestry Science & Technology, 2014, 41 (1): 94- 97.
doi: 10.3969/j.issn.1003-5710.2014.01.018
|
|
张丽勍, 段可, 邹小花, 等. 2016. 胶孢炭疽菌侵染草莓的转录组学研究. 中国植物病理学会2016年学术年会论文集.
|
|
Zhang L Q, Duan K, Zou X H, et al. 2016. Transcriptomics study of Colletotrichum gloeosporioides infecting strawberry. Proceedings of the 2016 Annual Conference of the Chinese Society of Plant Pathology. [in Chinese]
|
|
Castrense S , Liugi M P , Piero F , et al. BUSCA: an integrative web server to predict subcellular localization of proteins. Nucleic Acids Research, 2018, 46 (W1): W459- W466.
doi: 10.1093/nar/gky320
|
|
Dangl J L , Horvat D M , Staskawicz B J , et al. Pivoting the plant immune system from dissection to deployment. Science, 2013, 341 (6147): 746- 751.
doi: 10.1126/science.1236011
|
|
Djamei A , Schipper K , Rabe F , et al. Metabolic priming by a secreted fungal effector. Nature, 2011, 478 (7369): 395- 398.
doi: 10.1038/nature10454
|
|
Emanuelsson O , Brunak S , Heijne G , et al. Locating proteins in the cell using TargetP, SignalP and related tools. Nature Protocols, 2007, 2 (4): 953- 971.
doi: 10.1038/nprot.2007.131
|
|
Emanuelsson O , Nielsen H , Brunak S , et al. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. Journal of Molecular Biology, 2000, 300 (4): 1005- 1016.
doi: 10.1006/jmbi.2000.3903
|
|
Halaouli S , Asther M , Sigoillot J C , et al. Fungal tyrosinases: new prospects in molecular characteristics, bioengineering and biotechnological applications. Journal of Applied Microbiology, 2006, 100 (2): 219- 232.
doi: 10.1111/j.1365-2672.2006.02866.x
|
|
Jana S , Peter N D , Donald M G , et al. Improved prediction of fungal effector proteins from secretomes with Effector P2.0. Molecular Plant Pathology, 2018, 19 (9): 2094- 2110.
doi: 10.1111/mpp.12682
|
|
Jannick D B , Nielsen H , von Heijne G , et al. Improved prediction of signal peptides: signal P3.0. Journal of Molecular Biology, 2004, 340 (4): 783- 795.
doi: 10.1016/j.jmb.2004.05.028
|
|
Jin Q , Dong H , Peng Y , et al. Application of cDNA array for studying the gene expression profile of mature appressoria of Magnaporthe grisea. Journal of Zhejiang University Science B: Biomedicine & Biotechnology, 2007, 8 (2): 88- 97.
|
|
Jones D A , Bertazzoni S , Turo C J , et al. Bioinformatic prediction of plant-pathogenicity effector proteins of fungi. Current Opinion in Microbiology, 2018, 46, 43- 49.
doi: 10.1016/j.mib.2018.01.017
|
|
Jones J D G , Dangl J L . The plant immune system. Nature, 2006, 444 (7117): 323- 329.
doi: 10.1038/nature05286
|
|
Jonge R D , Esse H P V , Kombrink A , et al. Conserved fungal LysM effector Ecp6 prevents chitin-triggered immunity in plants. Science, 2010, 329 (5994): 953- 955.
doi: 10.1126/science.1190859
|
|
Juncker A S , Willenbrock H , Von Heijne G , et al. Prediction of lipoprotein signal peptides in gram-negative bacteria. Protein Science, 2003, 12 (8): 1652- 1662.
doi: 10.1110/ps.0303703
|
|
Kanehisa M , Sato Y . KEGG mapper for inferring cellular functions from protein sequences. Protein Science, 2019, 29 (1): 28- 35.
|
|
Ke X , Yin Z , Song N , et al. Transcriptome profiling to identify genes involved in pathogenicity of Valsa mali on apple tree. Fungal Genetics and Biology, 2014, 68, 31- 38.
doi: 10.1016/j.fgb.2014.04.004
|
|
Kushalappa A C , Yogendra K N , Karre S . Plant innate immune response: qualitative and quantitative resistance. Critical Reviews in Plant Sciences, 2016, 35 (1): 38- 55.
doi: 10.1080/07352689.2016.1148980
|
|
Liang X F , Shang S P , Dong Q Y , et al. Transcriptomic analysis reveals candidate genes regulating development and host interactions of Colletotrichum fructicola. BMC Genomics, 2018, 19 (1): 557- 578.
doi: 10.1186/s12864-018-4934-0
|
|
Martin U , Alayne C , James S , et al. PHI-base: the pathogen-host interactions database. Nucleic Acids Research, 2019, 48 (D1): D613- D620.
|
|
Peng T , Orsborn K I , Orbach M J , et al. Proline-rich vaccine candidate antigen of Coccidioides immitis: conservation among isolates and differential expression with spherule maturation. Journal of Infectious Diseases, 1999, 179 (2): 518- 521.
doi: 10.1086/314604
|
|
Ramachandran S R , Yin C , Kud J , et al. Effectors from wheat rust fungi suppress multiple plant defense responses. Phytopathology, 2017, 107 (1): 75- 83.
doi: 10.1094/PHYTO-02-16-0083-R
|
|
Shang S P , Wang B , Zhang S , et al. A novel effector CfEC92 of Colletotrichum fructicola contributes to glomerella leaf spot virulence by suppressing plant defences at the early infection phase. Molecular Plant Pathology, 2020, 21 (7): 936- 950.
doi: 10.1111/mpp.12940
|
|
Sperschneider J , Dodds P N , Gardiner D M , et al. Advances and challenges in computational prediction of effectors from plant pathogenic fungi. PLoS Pathogens, 2015, 11 (5): e1004806.
doi: 10.1371/journal.ppat.1004806
|
|
Yan W , Wang Y . Trick or treat: microbial pathogens evolved apoplastic effectors modulating plant susceptibility to infection. Molecular Plant-Microbe Interactions, 2018, 31 (1): 6- 12.
doi: 10.1094/MPMI-07-17-0177-FI
|