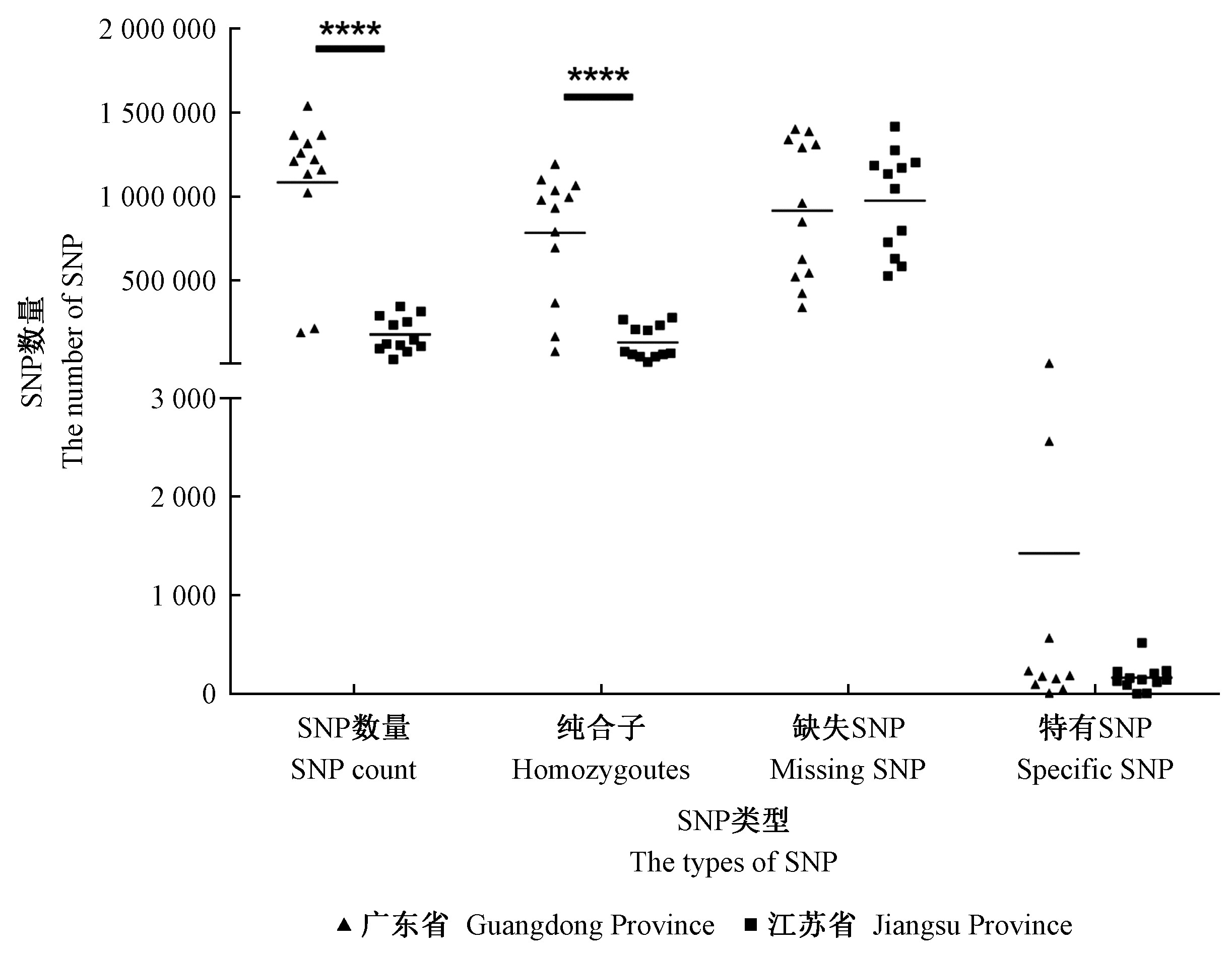
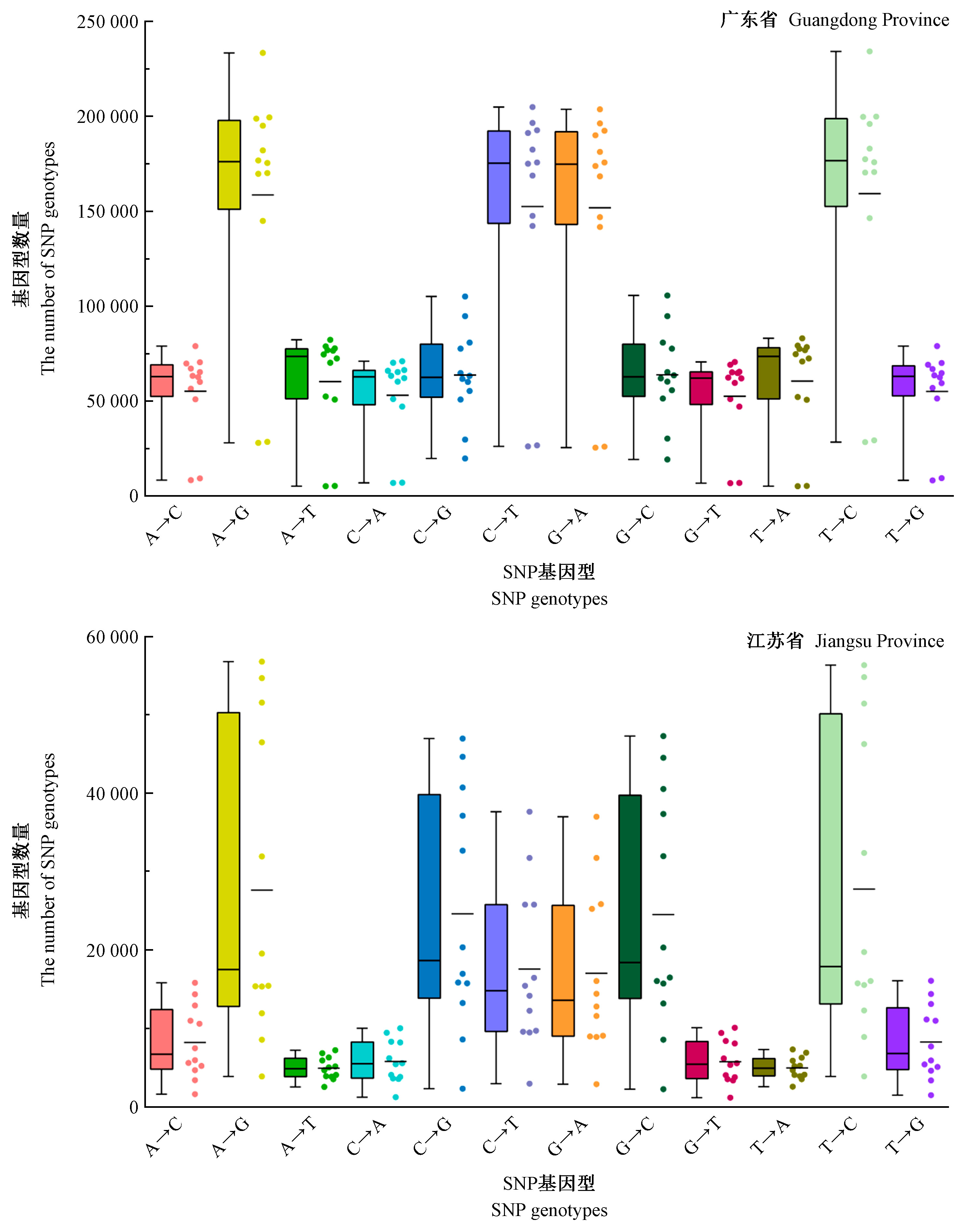
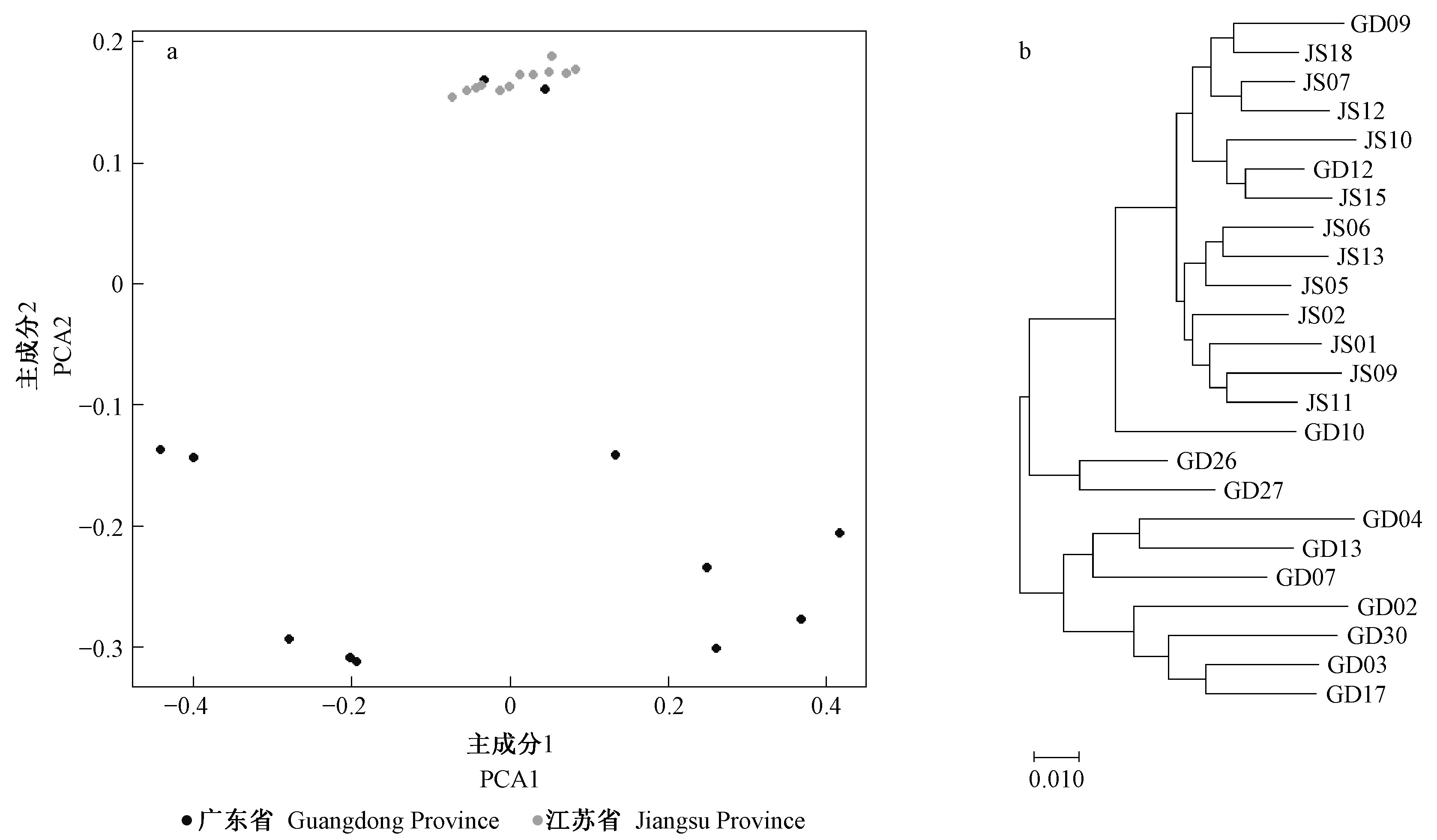
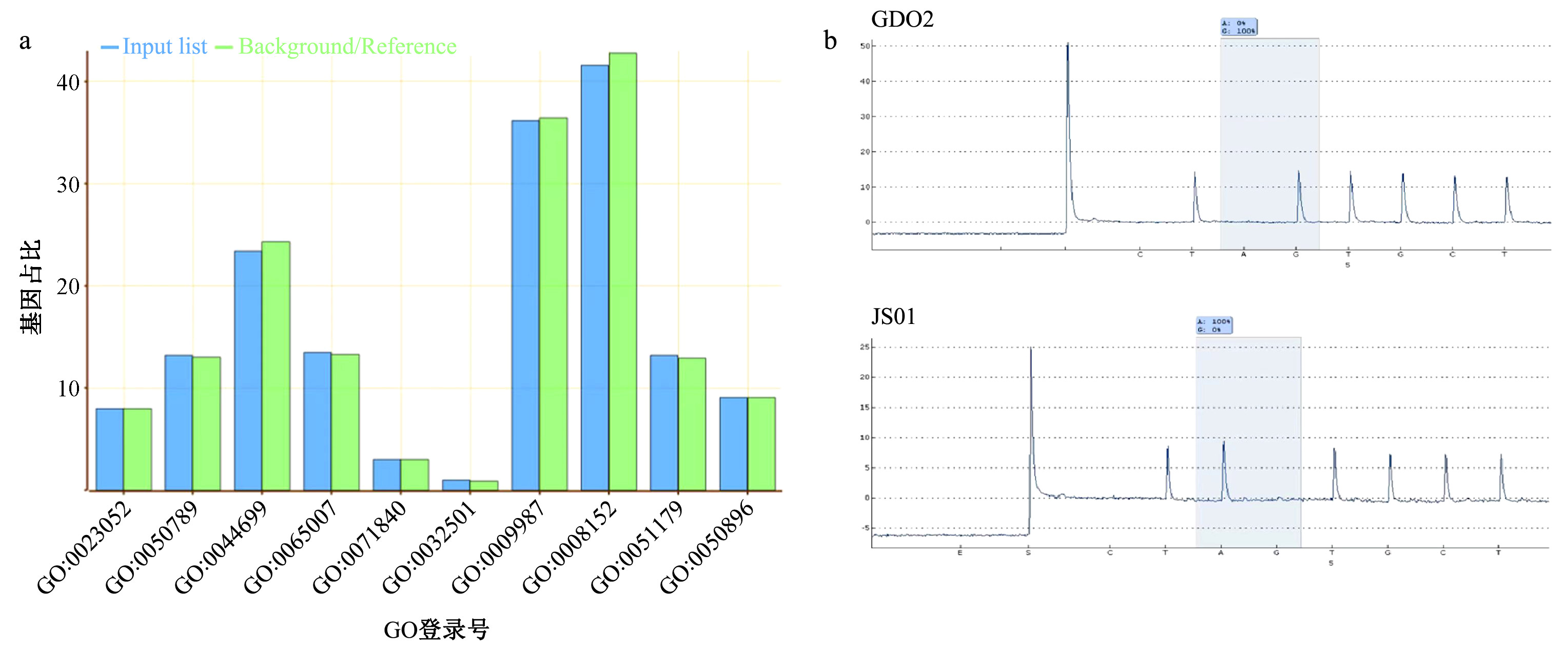
|
曾燕如, 黄敏仁, 王明庥. 一种新的分子标记——单核苷酸多态(SNP). 南京林业大学学报(自然科学版), 2003, 27 (3): 84- 88.
|
|
Ceng Y R , Huang M R , Wang M X . Single nucleotide polymorphisms, a new molecular marker. Journal of Nanjing Forestry University(Natural Sciences Edition), 2003, 27 (3): 84- 88.
|
|
陈凤毛. 2005. 松材线虫SCAR标记与分子检测技术. 南京: 南京林业大学.
|
|
Chen F M, 2005, SCAR marker and molecular detection technique of Bursaphelenchus xylophilus. Nanjing: Nanjing Forestry University [in Chinese]
|
|
陈秋玲, 高建明, 罗峰, 等. 分子标记技术在禾本科作物基因定位上的研究进展. 中国农学通报, 2010, 26 (9): 42- 48.
doi: 10.3969/j.issn.1007-7774.2010.09.015
|
|
Chen Q L , Gao J M , Luo F , et al. Research and development of molecular marker technologies for gene mapping of gramineous crops. Chinese Agricultural Science Bulletin, 2010, 26 (9): 42- 48.
doi: 10.3969/j.issn.1007-7774.2010.09.015
|
|
程瑚瑞, 林茂松, 黎伟强, 等. 南京黑松上发生的萎蔫线虫病. 森林病虫通讯, 1983, (4): 1- 5.
|
|
Cheng H R , Lin M S , Li W Q , et al. The nematode wilt disease on Pinus thunbergii in Nanjing. Forest Pest and Disease, 1983, (4): 1- 15.
|
|
黄金思, 奚晓桐, 丁晓磊, 等. 基于SNP标记的广东省松材线虫种群分化研究. 南京林业大学学报(自然科学版), 2019, 43 (6): 25- 31.
|
|
Huang J S , Xi X T , Ding X L , et al. Study on the population differentiation of Bursaphelenchus xylophilus in Guangdong Province by SNP markers. Journal of Nanjing Forestry University(Natural Sciences Edition), 2019, 43 (6): 25- 31.
|
|
黄文达, 赵学勇, 赵昕, 等. 分子标记在种群遗传学研究中的应用. 草业科学, 2010, 27 (11): 115- 120.
doi: 10.3969/j.issn.1001-0629.2010.11.018
|
|
Huang W D , Zhao X Y , Zhao X , et al. Application of molecular markers in population genetics. Pratacultural Science, 2010, 27 (11): 115- 120.
doi: 10.3969/j.issn.1001-0629.2010.11.018
|
|
黄族豪, 刘迺发. 种群遗传学研究进展. 安徽农业科学, 2008, 36 (31): 13490- 13491.
doi: 10.3969/j.issn.0517-6611.2008.31.007
|
|
Huang Z H , Liu Q F . Advances in population genetic. Journal of Anhui Agricultural Sciences, 2008, 36 (31): 13490- 13491.
doi: 10.3969/j.issn.0517-6611.2008.31.007
|
|
潘宏阳, 叶建仁, 吴小芹. 中国松材线虫病空间分布格局. 生态学报, 2009, 29 (8): 4325- 4331.
|
|
Pan H Y , Ye J R , Wu X Q . Spatial distribution patterns of China. Acta Ecologica Sinica, 2009, 29 (8): 4325- 4331.
|
|
潘佳亮, 姚翰文, 董瀛谦, 等. 2019年全国松材线虫病疫情分析. 中国森林病虫, 2020, 40 (1): 32- 37.
|
|
Pan J L , Yao H W , Dong Y Q , et al. Analysis of the epidemic situation of pine wilt disease in China in 2019. Forest Pest and Disease, 2020, 40 (1): 32- 37.
|
|
StoneG N, AtkinsonR J, BrownG, 等. 分布区扩张的群体遗传学后果: 类型与过程, 以栎瘿蜂为模型系统. 生物多样性, 2002, 10 (1): 80- 97.
|
|
Stone G N , Atkinson R J , Brown G , et al. The population genetic consequences of range expansion: a review of pattern and process, and the value of oak gallwasps as a model system,. Biodiversity Science, 2002, 10 (1): 80- 97.
|
|
谢丙炎, 成新跃, 石娟, 等. 松材线虫入侵种群形成与扩张机制——国家重点基础研究发展计划"农林危险生物入侵机理与控制基础研究"进展. 中国科学(C辑: 生命科学), 2009, 39 (4): 333- 341.
|
|
Xie B Y , Cheng X Y , Shi J , et al. The formation and expansion mechanism of invasion species Bursaphelenchus xylophilus——Key Project of National Fundamental Research and Development Programme" Basic study on dangerous species' invasion mechanism and control in agriculture and forestry ". Scientia Sinica (Vitae), 2009, 39 (4): 333- 341.
|
|
谢辉. 植物线虫分类学. 合肥: 安徽科学技术出版社, 2000: 186- 188.
|
|
Xie H . Taxonomy of plant nematodes. Hefei: Anhui Science and Technology Press, 2000: 186- 188.
|
|
许峻荣, 吴小芹, 刘云, 等. 基于松材线虫全基因组序列的SSR标记开发. 南京林业大学学报(自然科学版), 2014, 38 (2): 36- 42.
|
|
Xu J R , Wu X Q , Liu Y , et al. Development of simple sequence repeats base on pine wood nematode (Bursaphelenchus xylophilus) genom sequence. Journal of Nanjing Forestry University(Natural Sciences Edition), 2014, 38 (2): 36- 42.
|
|
叶建仁, 黄麟. 松材线虫病病原学研究的几个问题. 中国森林病虫, 2012, 31 (5): 13- 21.
|
|
Ye J R , Huang L . Some aspects of the pathogen of the pine wilt disease. Forest Pest and Disease, 2012, 31 (5): 13- 21.
|
|
叶建仁. 松材线虫病在中国的流行现状、防治技术与对策分析. 林业科学, 2019, 55 (9): 1- 10.
|
|
Ye J R . Epidemic status of pine wilt disease in China and its prevention and control techniques and counter measures. Scientia Silvae Sinicae, 2019, 55 (9): 1- 10.
|
|
Phillip A M , Gordon L , Robert K W , et al. SNPs in ecology, evolution and conservation. Trends in Ecology and Evolution, 2004, 19 (4): 208- 216.
|
|
Aikawa T , Kanzaki N , Maehara N . ITS-RFLP pattern of Bursaphelenchus xylophilus (Nematoda: Aphelenchoididae) does not reflect nematode virulence. Journal of Forest Research, 2013, 18 (4): 384- 388.
|
|
Bai H H , Guo X S , Narisu N , et al. Whole-genome sequencing of 175 mongolians uncovers population-specific genetic architecture and gene flow throughout North and East Asia. Nature Genetics, 2018, 50 (12): 1696- 1704.
|
|
Choi N R , Seo D , Jin S , et al. Discrimination of native chicken breeds using SNP markers selected from the 600K chip data. Journal of Animal Science, 2016, 94 (1): 91.
|
|
Figueiredo J , Simoes M J , Gomes P , et al. Assessment of the geographic origins of pinewood nematode isolates via single nucleotide polymorphism in effector genes. PloS ONE, 2013, 8 (12): e83542.
|
|
Gu J F , Wang J L , Braasch H , et al. morphological and molecular characterisation of mucronate isolates ('M' form) of Bursaphelenchus xylophilus (Nematoda: Aphelenchoididae). Russian Journal of Nematology, 2011, 19 (2): 103- 120.
|
|
Jung J , Han H , Ryu S H , et al. Amplified fragment length polymorphism analysis and genetic variation of the pinewood nematode Bursaphelenchus xylophilus in South Korea. Animal Cells and Systems, 2010, 14 (1): 31- 36.
|
|
Lander E S . The new genomics: global views of biology. Science, 1996, 274 (5287): 536- 539.
|
|
Nei M , maruyama T , Chakraborty R . The bottleneck effect and genetic variability in populations. Society for the Study of Evolution, 1975, 29 (1): 1- 10.
|
|
Woldegiorgis S T , Wang S B , He Y R , et al. Rice stress-resistant SNP database. Rice, 2019, 12 (1): 97.
|