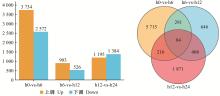
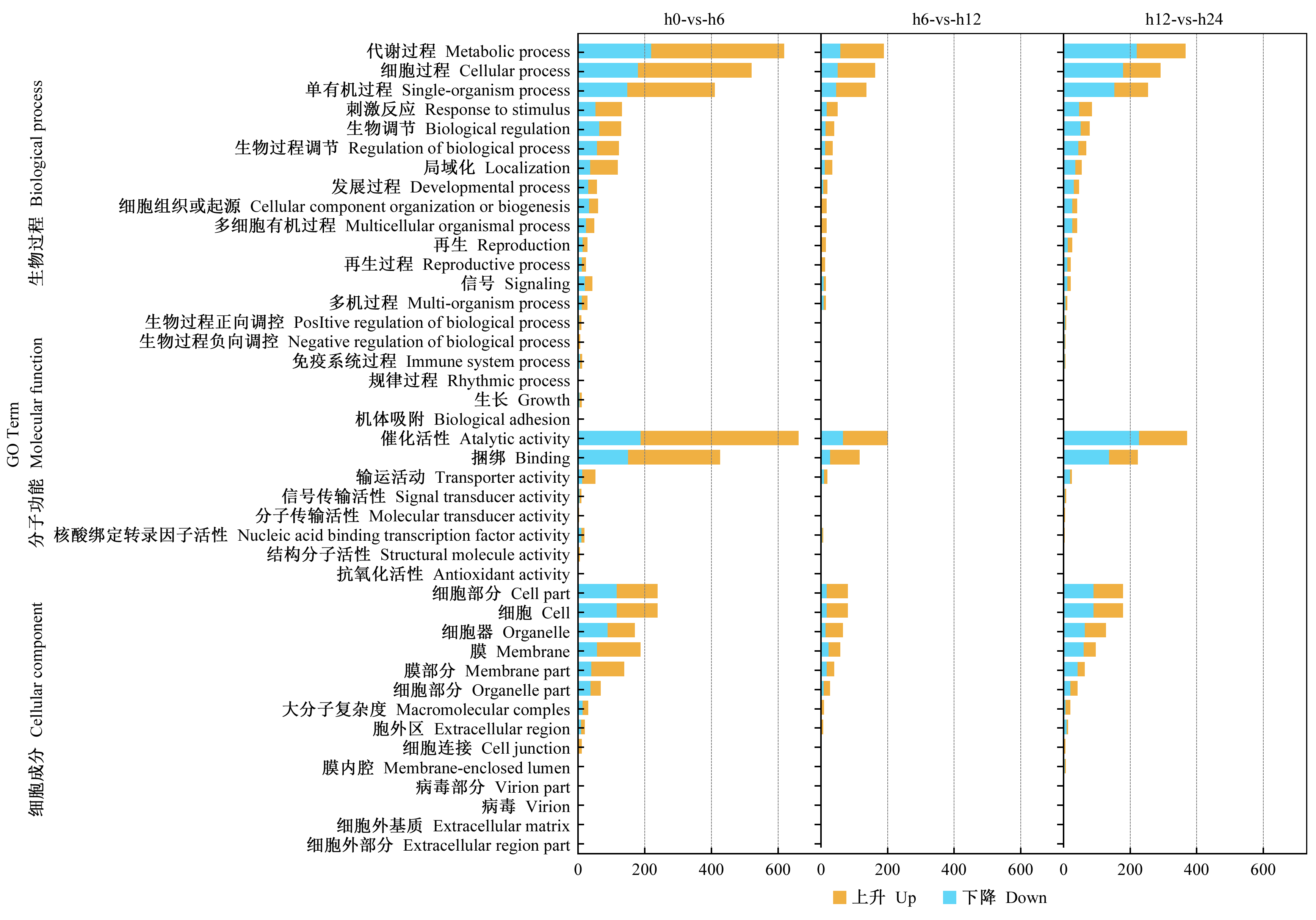
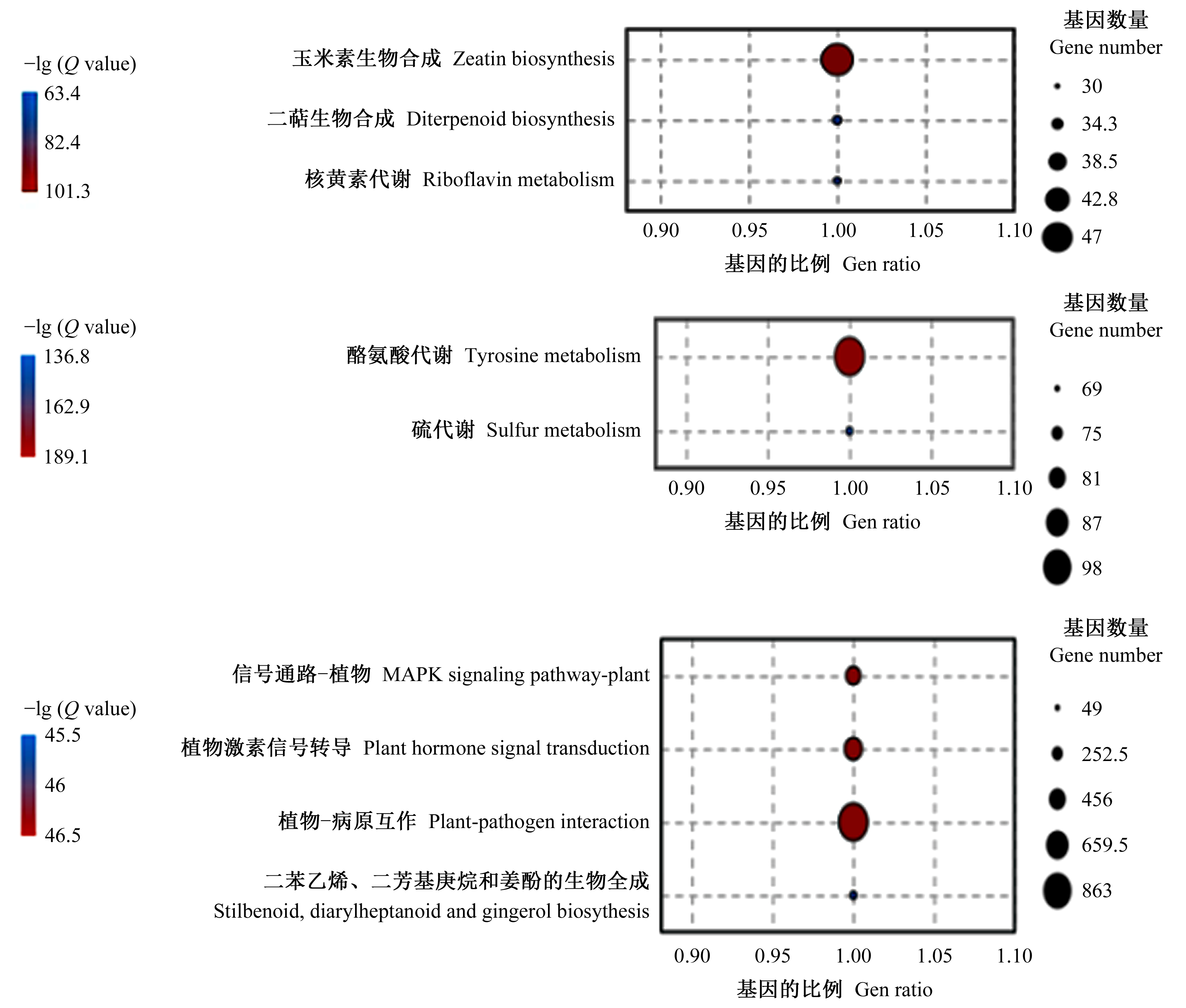
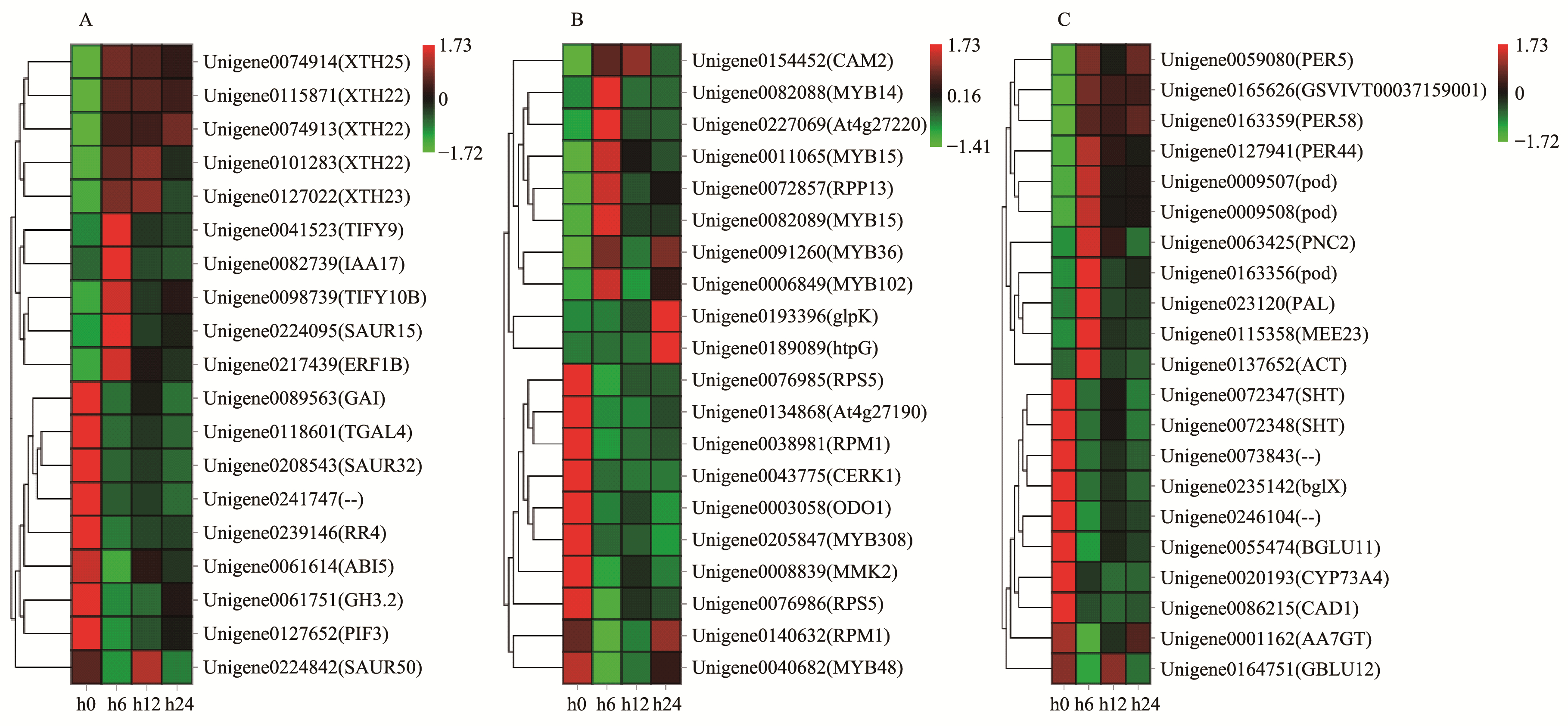
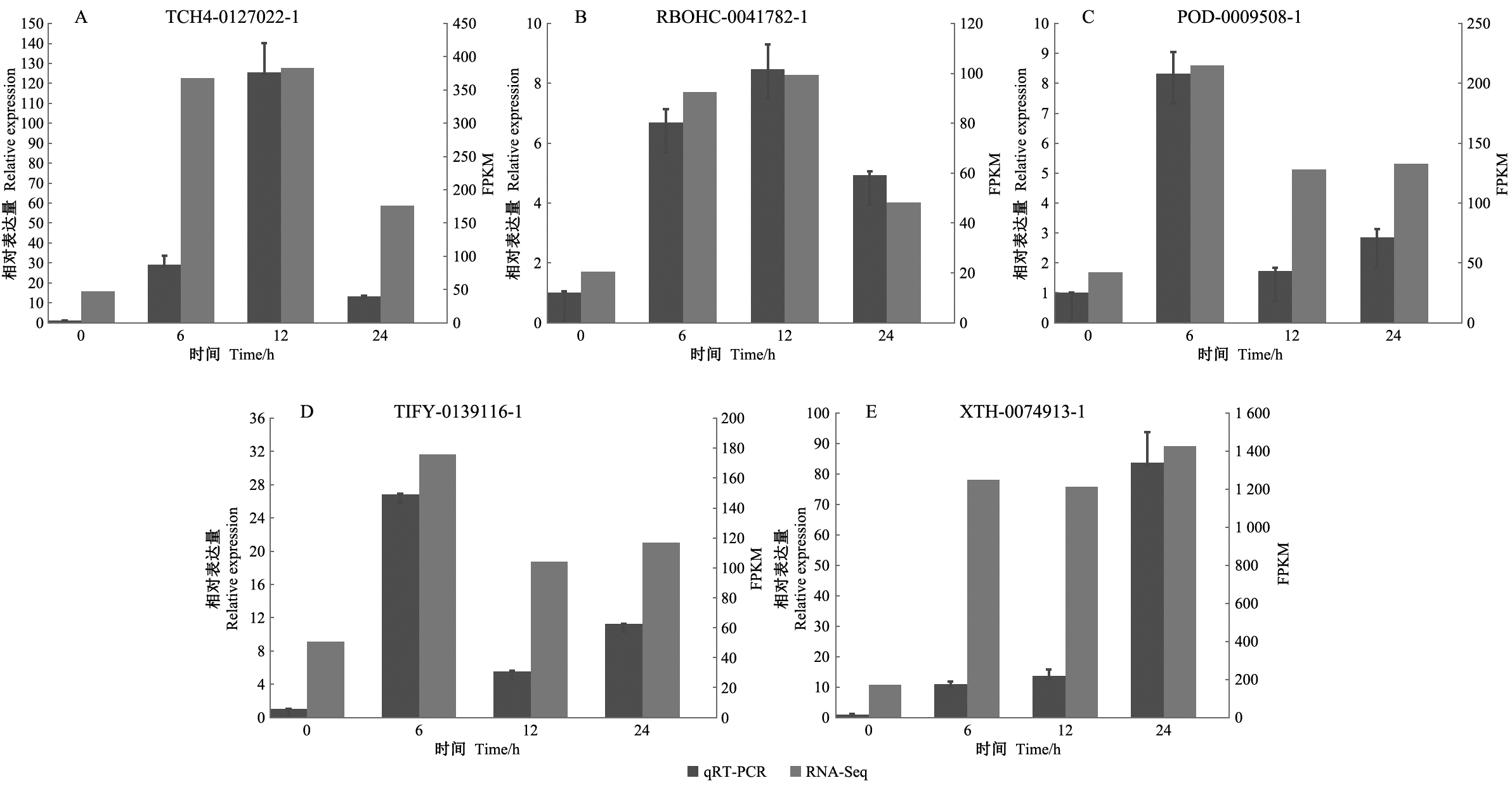
|
段龙飞, 慕小倩, 李文燕. 茉莉酸信号途径中转录抑制因子JAZ蛋白家族的分子进化分析. 植物学报, 2013, 48 (6): 623- 634.
|
|
Duan L F , Mu X Q , Li W Y . Molecular evolution analysis of the JAZ protein family of transcriptional suppressor in jasmonic acid signaling pathway. Acta Botanica Sinica, 2013, 48 (6): 623- 634.
|
|
张佳琦, 胡恒康, 徐川梅, 等. 核桃JrGA2ox基因的克隆、亚细胞定位及功能验证. 林业科学, 2019, 55 (2): 50- 60.
|
|
Zhang J Q , Hu H K , Xu C M , et al. Cloning, subcellular mapping and functional verification of JrGa2Ox gene in Walnut. Scientia Silvae Sinicae, 2019, 55 (2): 50- 60.
|
|
王恒煦, 刘泽平, 王志刚, 等. 3株芽孢杆菌在水稻根际定殖促生及其在土壤中的存活. 生态与农村环境学报, 2019, 35 (7): 892- 899.
|
|
Wang H X , Liu Z P , Wang Z G , et al. 3 strains of Bacillus colonize and promote growth in rice rhizosphere and their survival in soil. Journal of Ecology and Rural Environment, 2019, 35 (7): 892- 899.
|
|
Banerjee A , Roychoudhury A . WRKY proteins: signaling and regulation of expression during abiotic stress responses. The Scientific World Journal, 2015, 225 (2): 807560- 807561.
doi: 10.1155/2015/807560
|
|
Bechinger C . Optical measurements of invasive forces exerted by appressoria of a plant pathogenic fungus. Science, 1999, 285 (5435): 1896- 1899.
doi: 10.1126/science.285.5435.1896
|
|
Bulgarelli D , Rott M , Schlaeppi K , et al. Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature, 2012, 488 (7409): 91- 95.
doi: 10.1038/nature11336
|
|
Dominik K G , Eric van der G , Thomas R . Phytoalexin transgenics in crop protection-fairy tale with a happy end?. Plant Science, 2012, 195 (94): 54- 70.
|
|
Drogue B , Sanguin H , Chamam A , et al. Plant root transcriptome profiling reveals a strain-dependent response during Azospirillum-rice cooperation. Frontiers in Plant Science, 2014, 5 (10): 607- 608.
doi: 10.3389/fpls.2014.00607
|
|
Galambos N , Compant S , Moretto M , et al. Humic acid enhances the growth of tomato promoted by endophytic bacterial strains through the activation of Hormone-, Growth-, and Transcription-Related processes. Frontiers in Plant Science, 2020, 11 (21): 1437- 1438.
|
|
Irizarry I , White J . Bacillus amyloliquefaciens alters gene expression, ROS production, and lignin synthesis in cotton seedling roots. Journal of Applied Microbiology, 2018, 124 (6): 1589- 1603.
doi: 10.1111/jam.13744
|
|
Ishihama N , Yoshioka H . Post-translational regulation of WRKY transcription factors in plant immunity. Current Opinion in Plant Biology, 2012, 15 (4): 431- 437.
doi: 10.1016/j.pbi.2012.02.003
|
|
Johnson C , Arias B J . Salicylic acid and NPR1 induce the recruitment of trans-activating TGA factors to a defense gene promoter in Arabidopsis. Plant Cell, 2003, 15 (8): 1846- 1858.
doi: 10.1105/tpc.012211
|
|
King E , Wallner A , Rimbault I , et al. Monitoring of rice transcriptional responses to contrasted colonizing patterns of Phytobeneficial burkholderia s. l. reveals a temporal shift in JA systemic response. Frontiers in Plant Science, 2019, 10 (3): 1141- 1141.
doi: 10.3389/fpls.2019.01141
|
|
Langmead B , Salzberg S L . Fast gapped-read alignment with Bowtie 2. Nature Methods, 2012, 10 (4): 357- 358.
doi: 10.1038/nmeth.1923
|
|
Macho A P , Zipfel C . Plant PRRs and the activation of innate immune signaling. Molecular Cell, 2014, 54 (2): 263- 272.
doi: 10.1016/j.molcel.2014.03.028
|
|
Naoumkina M A , Zhao Q , Gallego-giraldo L , et al. Genome-wide analysis of phenylpropanoid defence pathways. Molecular Plant Pathology, 2010, 11 (6): 829- 846.
|
|
Pandey S P , Smssich I E . The role of WRKY transcription factors in plant immunity. Plant Physiology, 2009, 150 (4): 1648- 1655.
doi: 10.1104/pp.109.138990
|
|
Pankievicz V C , Camilios-Neto D , Bonato P , et al. RNA-seq transcriptional profiling of Herbaspirillum seropedicae colonizing wheat (Triticum aestivum) roots. Plant Molecular Biology, 2016, 90 (11): 589- 603.
|
|
Pertea G , Huang X , Liang F , et al. TIGR gene indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics, 2003, 19 (5): 651- 652.
doi: 10.1093/bioinformatics/btg034
|
|
Pinski A , Betekhtin A , Hupert-Kocurek K , et al. Defining the fenetic basis of plant-endophytic bacteria interactions. International Journal of Molecular Sciences, 2019, 28 (4): 1947- 1947.
doi: 10.3390/ijms20081947
|
|
Saenz-Mata J , Berenice Salazar-Badillo F , Francisco Jimenez-Bremont J , et al. Transcriptional regulation of Arabidopsis Thaliana WRKY fenes under interaction with beneficial fungus Trichoderma atroviride. Acta Physiologia Plantarum, 2014, 36 (5): 1085- 1093.
doi: 10.1007/s11738-013-1483-7
|
|
Sheibani-Tezerji R , Rattei T , Sessitsch A , et al. Transcriptome profiling of the endophyte Burkholderia phytofirmans PsJN indicates sensing of the plant environment and drought stress. MBIO, 2015, 6 (21): 14- 15.
doi: 10.1128/mBio.00621-15
|
|
Shidore T , Dinse T , Ohrlein J , et al. Transcriptomic analysis of responses to exudates reveal genes required for rhizosphere competence of the endophyte Azoarcus sp. strain BH72. Environmental Microbiology, 2012, 14 (5): 2775- 2787.
|
|
Singh N , Pandey G K . Calcium signatures and signal transduction schemes during microbe interactions in Arabidopsis Thaliana. Journal of Plant Biochemistry and Biotechnology, 2020, 29 (4): 675- 686.
doi: 10.1007/s13562-020-00604-6
|
|
Straub D , Yang H , Liu Y , et al. Root ethylene signalling is involved in Miscanthus sinensis growth promotion by the bacterial endophyte Herbaspirillum frisingense GSF30T. Journal of Experimental Botany, 2013, 64 (14): 4603- 4615.
doi: 10.1093/jxb/ert276
|
|
Stringlis I A , Yu K , Feussner K , et al. MYB72-dependent coumarin exudation shapes root microbiome assembly to promote plant health. Proceedings of the National Academy of Sciences of the United States of America, 2018, 115 (22): E5213- E5222.
doi: 10.1073/pnas.1722335115
|
|
Tian W , Hou C , Ren Z , et al. A calmodulin-gated calcium channel links pathogen patterns to plant immunity. Nature, 2019, 572 (7767): 131- 135.
doi: 10.1038/s41586-019-1413-y
|
|
Vogt T . Phenylpropanoid biosynthesis. Molecular Plant, 2010, 3 (2): 20- 25.
doi: 10.1093/mp/ssp106
|
|
Xu J X , Li Z Y , Lv X , et al. Isolation and characterization of Bacillus Subtilis strain 1-L-29, an endophytic bacteria from Camellia oleifera with antimicrobial activity and efficient plant-root colonization. PLoS ONE, 2020, 15 (4): e0232096.
doi: 10.1371/journal.pone.0232096
|
|
Yi Y , De Jong A , Frenzel E , et al. Comparative transcriptomics of Bacillus mycoides strains in response to potato-root exudates reveals different genetic adaptation of endophytic and soil isolates. Front Microbiol, 2017, 8 (13): 1487.
|
|
Yimer H Z , Nahar K , Kyndt T , et al. Gibberellin antagonizes jasmonate-induced defense against Meloidogyne graminicola in Rice. New Phytologist, 2018, 218 (2): 646- 660.
doi: 10.1111/nph.15046
|
|
Yuan P , Jauregui E , Du L , et al. Calcium signatures and signaling events orchestrate plant-microbe interactions. Current Opinion in Plant Biology, 2017, 38 (1): 173- 183.
|